

Об авторе

Андрей Степанович Брюховецкий — доктор медицинских наук, профессор, ветеран Министерства обороны РФ, полковник медицинской службы в запасе. В настоящее время является главным научным сотрудником лаборатории молекулярной и клеточной нейробиологии Школы биомедицины Дальневосточного федерального университета, генеральным директором Клиники восстановительной интервенционной неврологии и терапии «НейроВита», ведущим научным сотрудником Научно-исследовательского отдела Центральной клинической больницы Российской академии наук (РАН). Врач-невролог высшей категории, вице-президент Международной ассоциации нейровосстановления (International Association of Neurorestoratology), член редколлегии ряда научных журналов: «Гены и клетки», Journal of Translational Neuroscience and Clinics, Journal of Neurorestoratology. Применением клеточных препаратов в клинике занимается с 1989 г. в рамках программ Министерства обороны РФ. С 1996 по 2002 г. руководил лабораторией высоких технологий НИИ трансплантологии и искусственных органов им. В. И. Шумакова Минздрава РФ. С 2003 по 2013 г. был координатором научной отраслевой программы РАМН «Новые клеточные технологии — медицине». С 2002 по 2006 г. возглавлял кафедру клеточной восстановительной медицины ГОУ ВПО «Российский государственный медицинский университет» им Н. И. Пирогова. С 2012 по 2015 г. — руководитель Центра биомедицинских технологий ФГБУ «Федеральный научно-клинический центр» ФМБА России. Автор 205 публикаций в рецензируемых российских и международных научных журналах, 10 научных монографий в области регенеративной медицины, неврологии, онкологии и 15 глав в коллективных монографиях. Автор 16 патентов РФ, 5 международных заявок PCT и патента США.
E-mail: neurovita-as@mail.ru

Предисловие
Боковой амиотрофический склероз (БАС) — одно из самых тяжелых заболеваний нервной системы у человека. Для любого врача-невролога или нейрофизиолога встреча с больным БАС это всегда испытание, неловкое положение, а порой и отчаяние от своего бессилия и врачебной несостоятельности. Поставить этот страшный диагноз — большая ответственность для невролога, потому что, по сути, это смертный приговор, и врач должен сообщить об этом пациенту, найти нужные слова для него и его родственников, чтобы сгладить удар трагедии, по возможности предотвратить суицид и другие необдуманные действия, не покривив при этом душой.
На первый взгляд диагностика БАС не вызывает затруднений, так как заболевание имеет характерную клиническую картину и патогномоничные изменения на электронейромиографии (ЭНМГ), обусловленные поражением мотонейронов передних рогов спинного мозга. Если, будучи неврологом или специалистом в области нейронаук, вы не разбираетесь в тонкостях ЭНМГ, вам на помощь придет опытный нейрофизиолог, который сделает это исследование и поставит правильный диагноз за вас. Клиническая манифестация БАС возникает только тогда, когда у больного погибла подавляющая часть мотонейронов (по разным данным, до 80% и более). Но даже если представить, что нам удалось диагностировать БАС на ранней стадии, все равно помочь больному мы не сможем. Как и во времена Шарко, сегодня, в конце второй декады XXI в., эффективного лечения этого заболевания не существует.
Что же сегодня мы знаем о данной болезни? Да почти все, и в то же время почти ничего! Это парадоксальная ситуация. Впервые клиническую картину этого заболевания в 1869 г. описали французский исследователь Жан-Мартен Шарко и его ученик Аликс Жоффруа, а уже в 1871 г. Шарко выделил эту болезнь как самостоятельное нервное заболевание на основании описанного им симптомокомплекса фасцикуляций и атрофии мышц языка и конечностей, наличия спастических парезов и параличей, в том числе бульбарного паралича, и обнаружения при аутопсии склерозирования и атрофии клеток передних рогов спинного мозга. Главной научной заслугой Шарко было то, что он первый сопоставил найденные на аутопсии изменения в передних рогах, которые он обозначил как «склероз», с типичной клинической картиной болезни и дал соответствующее название этой, как он считал, «спинальной» болезни. Болезнь также еще называли «мышечной сухоткой», о которой в 1885 г. русский ученый К. В. Рот написал в своей известнейшей одноименной монографии. Он описал в ней несколько случаев БАС и также обнаружил патологические изменения в передних рогах и перерождение пирамидных путей в спинном и продолговатом мозге у этих пациентов. Учение о боковом амиотрофическом склерозе возникло значительно позже, уже в самом конце XIX и начале XX в., как новый научный взгляд на полученные рядом исследователей гистопатологические факты, свидетельствующие о тяжелом системном нейродегенеративном органическом дефекте в центральной нервной системе с последующим разрушением нервно-мышечного аппарата, приводящим к быстрой смерти пациента вследствие нарушения дыхания. Основную веху в формировании нового научного мировоззрения на эту болезнь заложил еще в 1883 г. русский врач А. Я. Кожевников, который показал, что БАС это не только поражение спинного мозга, как утверждал Шарко, а болезнь, связанная с перерождением всего пирамидного тракта. Она не ограничивается только спинным и продолговатым мозгом, а вовлекает в патологический процесс варолиев мост, ножки мозга, внутреннюю капсулу и белое вещество полушарий вплоть до среднего и верхнего отдела передней центральной извилины. Именно А. Я. Кожевников сделал революционные открытия и впервые в мире доказал вовлечение всего пирамидного тракта в патологический процесс при БАС. И. П. Мержеевский и А. Ф. Эрлицкий в 1883 г., P. Marie в 1885 г., В. А. Муратов в 1889 г., Hoche в 1897 г. и многие другие отечественные и зарубежные исследователи описали самые различные дегенеративные повреждения головного и спинного мозга — от глазодвигательных нервов до передних, боковых и задних столбов спинного мозга и даже мягкой мозговой оболочки (Czylarz u.Marburg: Beitrag zur Histologie und Pathogenese der S.L.A. Z. klin. Med., 1901). Сегодня эти морфофункциональные представления об объеме поражения нервной ткани при БАС значительно расширены и систематизированы, но полного понимания этиопатогенеза БАС, равно как и патогенетического лечения как не было, так и нет.
Современные геномные, транскриптомные, протеомные, метаболомные, секретомные и прочие «омные» исследования при БАС привели нас к пониманию определенных генетических и молекулярных механизмов, лежащих в основе этой болезни. Однако все эти знания носят, скорее, описательный характер и пока не позволяют приступить к разработке эффективного патогенетического лечения этого смертельного заболевания. Новые научные данные о БАС, включающие результаты экспериментального и математического моделирования и глобального анализа электронных баз данных генома и транскриптома, выполненного в рамках глобального проекта «Геном человека», так и остались отдельными компонентами большого и непонятного «паззла» под названием БАС. Не найдено главное системообразующее начало, которое могло бы объединить все известные научные факты в единую картину, позволяющую понять природу этой смертельной болезни и предложить эффективное лечение. Мы летаем в космос, разговариваем друг с другом, находясь на противоположных точках Земного шара или даже в космическом пространстве, делаем уникальные микрохирургические операции, трансплантируем органы и ткани, создаем уникальные «умные» лекарственные препараты, обладающие адресным действием, но мы абсолютно бессильны перед БАС, как и перед целым рядом подобных ему нейродегенеративных заболеваний (болезнь Альцгеймера, БАС-деменции, болезнь Паркинсона и пр.). Современная неврология от безысходности занимается созданием стратегий паллиативной помощи этим обреченным людям, осознавая свое бессилие перед быстрым и фатальным концом этой болезни. Для кардинального решения проблемы БАС нам необходим некий интегральный стержень, на который можно будет нанизать колоссальное количество разрозненной информации, сопоставить все эти данные в некой единой системе координат и увидеть «свет в конце туннеля».
Представленная вашему вниманию книга ни много ни мало претендует на то, чтобы дать полноценное системное представление о БАС и стать первым «лучом света» в беспроглядной мгле негативных прогнозов и паллиативных клинических подходов. Можно по-разному относиться к попытке систематизации знаний о БАС, предпринятой автором, но в одном, думаю, ему не сможет отказать ни один читатель: в оригинальности подхода, низвергающего представления классической неврологии о патоморфологии и патофизиологии БАС.
Автор ищет причину болезни не в очередном патоспецифическом белке, аккумулирующемся в мотонейронах и вызывающем его дегенерацию, а пытается объяснить патогенез БАС через системные механизмы нарушения иммунного ответа. Вопреки мировому тренду уходя от анализа локальных повреждений мотонейрона, автор пытается мотивированно объяснить аутоиммунный характер этого заболевания на примере аутоагрессии клонов иммунных клеток, возникших из патологической гемопоэтической стволовой клетки (ГСК). На самом деле причин первичного и вторичного поражения нервной ткани при БАС известно действительно много. За последние полтора века предлагались десятки противоречивых теорий возникновения БАС, от инфекционно-токсической и вирусной до врожденной слабости или генетической неполноценности нервной ткани. Анализируя существующие теории, автор пытается с позиций фундаментальной науки обосновать аутоиммунную теорию возникновения болезни, которая, по его мнению, является следствием системных нарушений на уровне генома и протеома ГСК костного мозга у пациента. Автор убежден, что этиологические факторы болезни формируют геномные, транскриптомные и протеомные изменения в биоинформационной структуре ГСК и определяют модификацию ее секретома, имеющего важнейшее значение в регуляции всей иерархии потомков аутологичной ГСК, в том числе и для цитотоксических ее потомков, опосредующих аутоагрессию. Автор вводит новый научный термин специфической недостаточности иммунной системы (НИС) и демонстрирует ее проявления при БАС. По мнению автора, диагностика НИС может быть осуществлена путем протеомного анализа мембранных маркеров аутологичных ГСК пациента. Удивительным образом автору удалось показать, что в семейных формах БАС патология ГСК может возникать раньше, чем повреждение мотонейронов. Этот феномен требует детальной проверки и уточнения, но если выявленные изменения протеомного профиля мембранных белков ГСК действительно окажутся специфичными для БАС на доклинической стадии, то нам открывается уникальная возможность ранней молекулярно-биологической диагностики и, возможно, даже профилактики данного заболевания. Очень интересно изучить этот феномен и при других нейродегенерациях, например при болезни Альцгеймера, где накопление патологических тау-белков в нейронах очень напоминает патологию при БАС.
Научные объяснения молекулярно-нацеленной терапии, представленные автором в последней главе монографии, мне показались довольно точными, основанными на объективных данных иммунного статуса больного и анализе нейроспецифических белков в крови и ликворе пациента. Они открывают новые перспективы таргетного лечения неизлечимых нейродегенеративных заболеваний. Опыт автора очень интересен, но требует тщательной проверки и, в случае его подтверждения, масштабирования при других заболеваниях.
Молекулярно-биологическая диагностика БАС, предложенная автором, открывает перспективы не только ранней диагностики, но и потенциального направления терапии. Поскольку, по мнению автора, в основе болезни лежит ГСК с патологически измененным протеомом, становится понятно, почему при БАС неэффективны аутологичная трансплантация костного мозга, гормонотерапия глюкокортикоидами и терапия блокаторами цитокиновых рецепторов, которые высокоэффективны при других аутоиммунных болезнях (системной красной волчанке, ревматоидном артрите или рассеянном склерозе). Если следовать логике автора, при БАС, в отличие от перечисленных выше заболеваний, иммуносупрессия аутореактивных клонов лимфоцитов не приводит к нормализации состава иммунокомпетентных клеток, поскольку измененные ГСК по-прежнему генерируют патологические клоны. Таким образом, по мнению автора, больным с БАС может помочь лишь трансплантация аллогенного костного мозга от иммуносовместимого донора, с последующим восстановлением поврежденных мотонейронов с помощью современных нейрорегенеративных технологий. Если предположение автора верно, то такая клеточно-лекарственная иммунотерапия БАС, целенаправленно воздействующая на ключевое звено патогенеза в виде измененной ГСК, обретает логику и здравый смысл. Так или иначе, но подход, основанный на персонализированном анализе протеома и развернутого иммунного статуса, представляется правильным и заслуживает доверие. В свете гипотезы о патологической ГСК, генерирующей цитотоксические клоны, повреждение мотонейрона при БАС автор считает не причиной, а следствием. Сама по себе эта точка зрения не является новой в неврологии, но в отличие от предыдущих исследователей, высказывающих ее, автор пытается найти логическое объяснение этого факта, а не просто констатирует его. Несомненно, предлагаемая гипотеза спорна и потребуются дополнительные исследования независимых коллективов, чтобы подтвердить или опровергнуть ее, однако в любом случае это — новый, альтернативный взгляд на фатальную болезнь, с которым интересно и полезно ознакомиться любому специалисту в области нейронаук.
В свете развития технологий редактирования генома с помощью CRISPR/Сas9, предлагаемая автором технология трансплантации алогенных ГСК может быть заменена на трансплантацию редактированных аутологичных ГСК. Более того, автором предлагается даже определенный сценарий такого редактирования, включающий исправление генов SOD1 и FUS.
Данная работа — итог многолетнего и кропотливого изучения бокового амиотрофического склероза автором и его коллегами и, при всей неоднозначности предлагаемых гипотез и подходов, эта интересная монография способствует расширению наших знаний о БАС и дает надежду на его успешную терапию в будущем. Пожелаем автору и нам всем дальнейших успехов в этой области и новых достижений.

Введение
Боковой амиотрофический склероз (БАС) — это тяжелое фатальное нейродегенеративное заболевание, для которого до настоящего времени не определена истинная причина и механизм формирования болезни и, к сожалению, не найдено реального способа излечения, а также не существует методов терапии даже для приостановки развития заболевания. Впервые в научной литературе БАС был описан 150 лет назад французским физиологом и врачом Жан-Мартеном Шарко (J.-M. Charcot), которому удалось связать наблюдаемую у пациентов спастичность и патологию в спинном мозге (Rowland et al., 2001, Овчинников, 2015; Брюховецкий и др., 2018). Он первый заявил о нозологической самостоятельности этой болезни в ряду нервных болезней. Шарко предложил название новой болезни: слово «амиотрофический» обозначает мышечную слабость и атрофию, слова «латеральный склероз» говорят о склеротизации передних и латеральных кортикоспинальных трактов, наблюдавшейся им у больных с БАС (Wijesekera et al., 2009). Подробное исследование БАС и разработки практических рекомендаций в нашей стране проводились выдающимися исследователями А. Я. Кожевниковым, Т. Л. Буниной, И. А. Завалишиным, М. Н. Захаровой (Бархатова и др., 1996; Бунина, 1962; Завалишин и др., 1999; Завалишин и др., 1990; Хондкариан и др., 1978; Скворцова и др., 2004, 2005; Овчинников, 2015; Брюховецкий и др., 2018).
Медиана выживаемости пациентов с диагностированным БАС составляет около 2–3 лет, в зависимости от конкретной формы и прогредиентности нервного заболевания. Очень редко (2–5%, по данным разных авторов) продолжительность жизни пациентов с БАС составляет менее 1 года, в 10–15% случаев это 7–10 лет. Описаны лишь несколько единичных случаев БАС, когда продолжительность жизни пациента составляла 25–30 лет. При этом все больные, прожившие более 20 лет, были абсолютно обездвиженными, парализованными тетраплегиками, но с полным сохранением продуктивности умственной деятельности и сохранностью интеллектуально-мнестических функций головного мозга. Наиболее ярким представителем последнего типа течения БАС является английский ученый-астроном профессор Стивен Хокинг, умерший в 2017 году, но проживший с БАС более 30 лет и активно занимавшийся фундаментальными научными исследованиями в области астрофизики. Но это больше исключение из правил, чем правило.
БАС сегодня — безусловно, летальная болезнь, которая очень быстро инвалидизирует пациента, резко ограничивает его возможности для самообслуживания, самостоятельности и жизнедеятельности и заканчивается внезапной смертью от удушья в результате паралича дыхательной мускулатуры. Тот человек, кто когда-нибудь видел, как умирают пациенты с БАС, обязательно подтвердит и скажет, что это одна из самых страшных и мучительных смертей, которая может быть у человека. В Библии написано, что болезни даются нам как испытания, и БАС — одно из самых страшных из всех существующих. Подобная смерть гораздо страшнее, чем смерть при раке или тяжелом сосудистом заболевании (инфаркте миокарда, инсульте), являющихся самыми частыми причинами смертности человечества в мире. При раке и целом ряде других злокачественных новообразований у пациента снижается уровень сознания до глубокого оглушения, сопора или комы в результате нарастающей раковой интоксикации или отека мозга, а при инсульте и инфаркте миокарда утрата сознания возникает в результате кардиогенного шока или отека (мозга или легких), так что человек спокойно уходит из жизни в бессознательном состоянии, не осознавая мучительного процесса расставания с жизнью. Смерть при БАС происходит при полном и ясном сознании, понимании и осознании всего происходящего самим пациентом. У больного, как правило, развивается паралич дыхания, и он понимает, что через несколько минут он умрет, и эти последние минуты его жизни самые страшные, когда сердце еще продолжает биться, а паралич дыхательного центра и дыхательной мускулатуры не позволяет сделать спасительный вдох. Конечно, больной может быть переведен на аппаратное искусственное дыхание (искусственную вентиляцию легких — ИВЛ) и на нем жить достаточно долго и мучительно. Однако официальная государственная медицина во всем мире не предлагает такого сценария помощи этим больным или формы лечения и поэтому не рекомендует переводить пациентов с БАС на ИВЛ. Подход простой: отсутствует действенный способ лечения, соответственно, гуманнее дать больному умереть. При этом официальная медицина не запрещает это делать родственникам, но только в 0,1% случаев родственники больных берут на себя такую ответственную миссию. В своей 30-летней неврологической врачебной практике мне пришлось видеть только четырех пациентов с БАС, родственники которых не смирились с таким исходом болезни и в течение многих лет вели борьбу за жизнь больных на аппарате ИВЛ.
Существуют ли терапевтические средства для эффективного лечения БАС в настоящее время? Ответ банален и трагичен. Нет, не существуют, так как нет понимания сущности данной болезни и ее причинно-следственных отношений. Насколько реально, даже с теоретических позиций, создать эффективное терапевтическое средство для лечения БАС? Абсолютно точно в рамках современной концепции «болезни моторного нейрона» терапевтического решения проблемы БАС нет. Волшебная пуля в виде лекарственного препарата, способного излечить больного с БАС, вряд ли будет создана в ближайшее время. Но вот новая методология понимания БАС как протеомного заболевания генома и эпигенома аутологичной гемопоэтической стволовой клетки (ГСК) человека позволяет по-новому посмотреть на возможность излечения от этой смертельной болезни и изменить взгляды на прогноз жизни у этих больных. Возможно, она приведет к разработке новой биомедицинской технологии, способной диагностировать болезнь на самой ранней стадии процесса, и, соответственно, в книге предлагается теоретическое обоснование лечения, способного остановить болезнь и даже полностью излечиться от этой болезни.
Почему мы не стали развивать это направление сами, а остановились на «половине дороги», на своих теоретических изысканиях? Наша команда работает в России на базах частных клиник, а не в государственных учреждениях, включенных в список Минздрава России. Другими словами, это связано с нововведениями и изменением законодательства нашей страны Минздравом России (приказ Минздрава России №875-н от 2018 г.) о запрете на манипуляции с ГСК, костным мозгом и проведения ТКМ в частных негосударственных организациях. Дальнейшие практические исследования в этом направлении в России будут преследоваться в уголовном порядке. Поэтому как законопослушные граждане мы остановили все работы в этом направлении. Но это не может запретить нам думать о тех возможностях и потенциале, которые открывают нам манипулирование ГСК и возможности аллогенной ТКМ или ТКМ после редактирования генома аутологичных ГСК.
Отсутствие хотя бы минимальных успехов в диагностике и лечении БАС связано со спецификой клинической манифестации данной болезни. Такая болезнь, как БАС, начавшись у человека, протекает очень длительное время бессимптомно и манифестирует клинически только тогда, когда общее морфологическое повреждение мотонейронов у человека достигает около 75–90%. Этот научный факт был подмечен еще самим Ж.-М. Шарко и подтвержден целой плеядой неврологов во всем мире. Если помните, то профессор Н. Н. Бурденко, великий отечественный нейрохирург, в 20-х гг. прошлого века подметил важный экспериментальный факт, заключающийся в том, что если у человека с повреждением спинного мозга сохранены более 10% мотонейронов в боковых столбах спинного мозга, то он способен ходить, даже не замечая этого дефекта. Так и при БАС, большая часть (до 90%) мотонейронов у больного погибает, когда заболевание еще ничем клинически не проявляется. Другими словами, асимптомность дебюта и скрытость течения БАС на ранних стадиях патологического дегенеративно-атрофического процесса в нервной и мышечной системах организма пациента приводят к тому, что клинические проявления заболевания возникают уже в случае глубокого склеротического органического и протеомного дефекта мотонейронов и других клеток нервной ткани в головном и спинном мозге человека. То есть тогда, когда обратного пути для нейрорегенерации нет и восстановление мотонейронов практически невозможно. Однако психологически манифестация неврологических симптомов БАС воспринимается заболевшим человеком как истинное начало его болезни, как возникновение болезни среди полного здоровья, «как гром среди ясного неба», то есть всегда неожиданно и трагично. Дебют болезни осознается пациентом и его родственниками только тогда, когда существующий органический дефект мотонейронов спинного и головного мозга при данном заболевании уже тотален и необратим. Неуклонно прогрессируя, долгое время болезнь развивается бессимптомно или малосимптомно, манифестация заболевания начинается, как правило, лавинообразно в виде нарастания неврологической симптоматики, инвалидизируя и разрушая моторную сферу пациента, уродуя атрофиями мышечную систему и нарушая естественную моторику движений. Мучительные фибриллярные подергивания (фибрилляции) отдельных групп мышц в разных участках тела становятся невыносимыми для больных, невротизируя их и приводя к астенизации и депрессии. В дегенеративный процесс постепенно и неуклонно вовлекается вся двигательная сфера пациента с формированием атрофий скелетных мышц и мышц гладкой мускулатуры органов на фоне массивной дегенерации мотонейронов, при полной сохранности интеллектуально-мнестической сферы у заболевшего. Именно поэтому в последние годы это заболевание стали чаще называть болезнью моторного нейрона (БМН). В первой главе монографии мы обсудим это название заболевания более обстоятельно.
Можно ли диагностировать повреждение мотонейронов на ранних стадиях заболевания? Возможно ли это хотя бы с теоретических позиций? Можно ли найти какие-то ранние маркеры диагностики БАС, когда еще сохранена большая часть мотонейронов человека? Можно ли на ранней стадии своевременно остановить дегенеративно-атрофический процесс в нервной ткани и опорно-двигательном аппарате? Мы попытаемся дать в этой монографии основные ответы на поставленные нами сложнейшие вопросы современной медицины вообще и неврологии в частности.
С современных позиций классической неврологии диагноз данного заболевания является отчасти клиническим, но в большей мере электронейромиографическим. То есть диагноз БАС должен быть обязательно подтвержден нейрофизиологическим поражением мотонейронов переднего рога спинного мозга и боковых столбов спинного мозга с использованием обычной электронейромиографии (ЭНМГ), игольчатой ЭНМГ и ЭНМГ с вызванными потенциалами. Мы считаем, что в данном раскладе ранняя диагностика БАС практически невозможна технически, так как повреждение в СМ должно быть значительным и нейрофизиологически значимым.
На сайте ALS Info (http://als-info.ru/10-krupnejshih-otkrytij-v-issledovanii-bas/) 24 октября 2017 г. были опубликованы 10 последних открытий в научных исследованиях о БАС за последние полтора года. Очевидно, что было совершено несколько важных научных открытий о БАС, достигнуты успехи в клинических испытаниях, созданы новые научные объединения и определены стратегические инициативы — все ради одной цели: как можно быстрее выяснить причины БАС и найти лекарство. Вот как выглядят эти 10 самых значимых научных фактов в области исследования БАС, которые дают ученым, врачам и пациентам надежду: 1. Подтверждено, что ген NEK-1 связан с наследственной формой БАС. В самом масштабном в истории изучения БАС исследовании семейных случаев заболевания участвовали более 80 ученых из 11 стран. Финансирование этого исследования стало возможно благодаря средствам, собранным во время проведения флешмоба Ice Bucket Challenge. Исследование было организовано и проведено проектом Project MinE, цель которого — собрать и расшифровать максимальное количество ДНК больных БАС и найти гены, ответственные за возникновение заболевания. 2. Одобрен новый препарат, незначительно замедляющий БАС. Управление по контролю за продуктами питания и лекарственными средствами США (FDA) разрешило применение препарата Эдаравон для терапии БАС. 3. ПЭТ-исследование пациентов с БАС. Команда ученых из департамента функциональной диагностики и визуальных методов исследования Научного клинического института неврологии в многопрофильной больнице Массачусетса во главе с доктором Наземом Атасси впервые провела ПЭТ-исследование организма человека с БАС, чтобы оценить воспалительные процессы в головном мозге. Позитронно-эмиссионная томография (ПЭТ) — активно развивающийся диагностический и исследовательский метод ядерной медицины. В основе этого метода лежит возможность при помощи специального детектирующего оборудования (ПЭТ-сканера) отслеживать распределение в организме биологически активных соединений, меченных позитрон-излучающими радиоизотопами. Считается, что показатель воспаления является важным визуальным биомаркером. Сейчас в исследовании принимает участие большое количество пациентов с БАС. 4. C9orf72 попал под прицел. При поддержке американской Ассоциации БАС (ALSA) доктор Аарон Гитлер и его команда исследователей выявили новую мишень для терапии мутаций гена C9orf72, чрезмерная экспрессия которого связана с наследственной формой БАС. 5. Генную терапию объединили с терапией стволовыми клетками. Ученые в больнице Cedars-Sinai в Лос-Анджелесе получили одобрение FDA на проведение клинического исследования по комбинации генной терапии и терапии стволовыми клетками. Ранее Ассоциация сертифицировала больницу как передовой научно-исследовательский центр по поиску лекарства от БАС. Клиника Cedars-Sinai соответствует самым строгим стандартам ALSA и в работе базируется на современном мультидисциплинарном подходе в уходе и оказании помощи пациентам. 6. Завершилась вторая фаза исследования NurOwn. Инновационная биотехнологическая компания Brainstorm, которая занимается использованием стволовых клеток (СК) в терапии неизлечимых заболеваний, объявила о начале проведения третьей фазы клинических испытаний в 2017 г. NurOwn — это платформа клеточной терапии, источником которой являются мезенхимальные клетки из костного мозга пациентов с БАС. Исследователи наделяют мезенхимальные клетки способностью продуцировать факторы роста для нейронов, которые являются своего рода питанием для клеток. Исследования на животных показали, что эти факторы роста обладают также значительным защитным потенциалом. 7. Обнаружены пять новых генов, ответственных за возникновение БАС. Это стало возможным благодаря суперкомпьютеру Watson компании IBM. Совместная работа IBM и Неврологического института Бэрроу в Фениксе показала, насколько структурирование и обработка больших данных, а также передовые компьютерные технологии могут ускорить процесс поиска лечения от БАС. 8. В США начала работу программа GTAC. GTAC (англ. Genomic Translation for ALS Care — Генетическая расшифровка для помощи при БАС) — это одна из самых крупных и высокотехнологичных медицинских программ по расшифровке генов, связанных с развитием БАС. В программу входят девять центров в университетах и клиниках по всей стране. Ассоциация БАС выделила 3,5 млн долларов из средств, полученных во время Ice Bucket Challenge, на развитие этой инициативы. Целью медицинской программы является исследование генетического материала и клинических проявлений у больных БАС и поиск общих закономерностей. В исследованиях участвуют более 1500 пациентов. 9. Завершена вторая стадия исследования препарата «Аримокломол». Исследование проводят ведущий центр исследования БАС университета Майами и фармацевтическая компания Orphazyme. «Аримокломол» исследуется для лечения быстро прогрессирующей наследственной формы БАС с мутацией гена SOD1. 10. Проекты ALS ONE и NeuroLINCS объявили о начале совместной работы. ALS ONE объединяет четыре крупнейших центра по изучению БАС в США: Многопрофильный госпиталь в Массачусетсе, Институт развития терапии БАС (ALS TDI), медицинский факультет университета Массачусетса и Организацию по развитию ухода за людьми с БАС (CCALS). В новом совместном проекте исследователи будут анализировать данные, необходимые для более глубокого понимания работы нейронов и причин нейродегенеративных заболеваний.
В этой книге мы попытались предложить свою альтернативную авторскую концепцию ранней диагностики БАС, основанную на оценке молекулярно-биологических особенностей белков мембранной поверхности гемопоэтических стволовых (CD34+) клеток (ГСК) у пациентов. Наши собственные исследования показали, что в основе ранней диагностики БАС, как и рака и других злокачественных опухолей, лежит нозоспецифическая иммунная недостаточность, обусловленная геномной, транскриптомной и протеомной трансформацией собственных ГСК как родоначальниц всех клеток иммунной системы пациента. Мы установили нозоспецифические протеомные профили белковых маркеров мембранной клеточной поверхности ГСК при БАС и раке, а также показали их кардинальное отличие их друг от друга. Поэтому мы полагаем, что постоянное мониторирование протеомного профилирования мембранной поверхности ГСК у лиц, находящихся в группе риска, может позволить диагностировать предрасположенность к БАС, семейные формы БАС и осуществлять диагностику аутоиммунного и нейродегенеративного процесса на самых ранних этапах заболевания, когда имеет место только начало заболевания без клинических и ЭНМГ-признаков болезни. Также в этой монографии мы обобщили все данные о возможности применения различных типов стволовых клеток в лечении БАС и представили собственное мнение об их эффективности при данной патологии.
Если вы введете в интернете ключевые слова «стволовые клетки в лечении бокового амиотрофического склероза», то получите 75 млн результатов и ссылок на сайты, которые активно предлагают применение СК в лечении БАС, из которых несколько сотен тысяч сайтов обсуждают или осуждают применение СК при этой болезни. Я думаю, что на самом деле большинство людей, которые писали эти сайты, никогда не занимались клеточной терапией в принципе, ввиду отсутствия у них стандартизированных продуктов из СК, так как их изготовление является очень трудоемким и высокотехнологическим процессом, который способна освоить только очень хорошо оснащенная лаборатория молекулярной и клеточной биологии. Такие лаборатории в нашей стране и за рубежом можно перечислить по пальцам. Поэтому очевидно, что авторы большинства сайтов просто переписывают информацию друг у друга и теоретизируют в публицистическом жанре. Те же ученые и исследователи, кто обсуждает недостатки и ужасы применения СК при БАС, лишь пытаются привлечь к себе пациентов на очередном «новомодном» методе реабилитационного лечения БАС, абсолютно не веря в его эффективность и не понимая смысла данного лечения. Однако сегодня следует признать, что в интернете большая часть сайтов о применении стволовых клеток при БАС дает позитивную и многообещающую информацию. Еще 10 лет назад написать статью о терапии БАС стволовыми клетками было просто дурным тоном, и чаще всего эти статьи не принимались редакциями большинства рейтинговых рецензируемых журналов как псевдонаучные.
Почему множество врачей и ученых считают, что именно клеточные технологии способны продлить жизнь пациента с БАС и добиться стабилизации его состояния? Это связано с безысходностью ситуации с БАС, а также с большими надеждами, основанными на целой серии зарубежных публикаций из стран Европы и США об эффективности клеточной терапии при БАС. В итоге это сказалось на изменении отношения к терапии БАС с использованием клеточных продуктов в России.
Мой личный научный интерес к этой проблеме существует уже давно. Более 30 лет я занимаюсь применением клеточных препаратов при лечении нервных болезней, в том числе и при БАС, и не могу разделить ни первую, ни вторую точку зрения относительно применения СК при нейродегенеративных заболеваниях. Я абсолютно уверен и точно знаю, что сама по себе технология применения клеточных препаратов для лечения нейродегенеративного заболевания действительно работает, но только тогда, когда решены фундаментальные вопросы остановки патологического процесса и прогредиентности заболевания и преодолены молекулярно-генетические проблемы нозоспецифической гистосовместимости клеточных систем и донорской нервной ткани и реципиента. Попытаюсь пояснить свой собственный, и далеко не формальный интерес к этой проблеме, а также 30-летний опыт пока еще низкоэффективных попыток применения различных клеточных систем в эксперименте и клинике при БАС. Ответ на этот вопрос лежит в истории моего личного опыта применения клеточных препаратов для лечения БАС.
Впервые в моей клинической практике я применил клеточный препарат для лечения пациента с БАС 15 апреля 1993 г. Будучи старшим ординатором неврологического отделения 32 Центрального военно-морского клинического госпиталя Министерства обороны РФ, я лично осуществил первое в мире введение препарата фетальных нервных клеток в субарахноидальное пространство больного с БАС. Препарат был изготовлен в лаборатории клинической иммунологии ГУ НИИ акушерства, гинекологии и перинатологии РАН (Москва) по биотехнологиям руководителя этой лаборатории профессора Г. Т. Сухих, изготовителем препарата был кандидат медицинских наук А. Ю. Аникин. Мы так подробно останавливаемся на деталях, чтобы вспомнить пионеров этого уникального направления развития медицины. Но это было далеко не началом истории терапии клеточными препаратами при БАС.
В течение трех лет (с 1990 г.) в рамках закрытых программ по нейротрансплантации Министерства обороны РФ на базе НИИ трансплантологии и искусственных органов Минздрава РФ (директор академик РАН и РАМН В. И. Шумаков) нашей командой военных врачей отрабатывалось применение фетальных нервных клеток для лечения боевой травмы головного и спинного мозга. Технология клеточной терапии повреждений мозга сначала была изучена в эксперименте на крысах, собаках и телятах с огнестрельными и минно-взрывными ранениями на базе ГУ НИИ трансплантологии и искусственных органов Минздрава РФ и Российского университета дружбы народов им. Патриса Лумумбы. Когда были получены первые результаты трансплантации фетальных нервных клеток в поврежденный спинной мозг экспериментальных животных и показана возможность регенерации поврежденной нервной ткани, в качестве контроля были выполнены более 50 трансфузий данных клеток в спинномозговое пространство животных. Был зарегистрирован клинико-морфологический эффект отчетливой регенерации мозга, почти такой же, как и при прямой трансплантации клеток, непосредственно введенных в ткань СМ. Главной особенностью этих исследований был подмеченный нами экспериментальный факт восстановления дегенерировавших нервных волокон боковых столбов спинного мозга при спинномозговой травме. Позже мы подтвердили эти научные факты и у человека. Однако, завершив эксперименты на животных, мы никак не могли перейти к применению клеточных препаратов на людях. Раньше до нас никто и никогда в мире не применял интратекальное введение клеточных препаратов для лечения нервных болезней. И это притом, что тогда уже более 10–15 лет в спинномозговой канал вводились антибиотики и химиопрепараты, однако клеточную суспензию вводить интратекально было страшно и опасно. Тем более, что это касалось не онкологических больных, а пациентов неврологического профиля. Необходимо было быть уверенным в том, что после цитотрансфузии клеточного препарата в спинномозговой канал не будет тяжелых витальных осложнений, связанных с возможной окклюзией ликворных путей у человека введенным клеточным препаратом. Ученый совет и Этический комитет НИИ транспланталогии и искусственных органов Минздрава России, учитывая мишень этих клеток в виде боковых столбов спинного мозга, порекомендовали нам сделать модель нейродегенеративного заболевания на 30 животных с болезнью Паркинсона и 30 крысах с моделью типа БАС. Мы выполнили эту работу и увидели потрясающий научный феномен: все фетальные нервные клетки, стереотаксически введенные в нервную ткань здорового головного и спинного мозга животных, лизировались и уничтожались, а при наличии повреждений в мозге эти клетки встраивались в зоны повреждения и частично восстанавливали функцию. Но самое интересное явление отмечалось при трансфузии суспензии фетальных нервных клеток в ликворное пространство. Эти клетки формировали конгломераты и «прилипали» к мягкой мозговой оболочке ГМ и СМ, затем вокруг конгломерата клеток формировалась новая мягкая мозговая оболочка, и конгломерат клеток становился частью нервной ткани. В дальнейшем клетки из конгломерата мигрировали в зоны повреждения ГМ и СМ и восстанавливали функцию поврежденных зон ГМ и СМ. Подробный отчет о данной работе представлен в монографии А. С. Брюховецкого «Нейротрансплантация и тканевая инженерия мозга в лечении нервных болезней», вышедшей в свет в 2003 г. Поэтому, прежде чем начать применение этих клеточных систем у раненых бойцов нашего госпиталя, было решено проверить безопасность введения суспензии фетальных нервных клеток у человека на терминальных стадиях больных с БАС, где риск был минимальным. Первым пациентом, получившим фетальные нервные клетки, стал больной мичман запаса Кар-в, 49 лет, с бульбарной формой БАС. Клетки для трансфузии в спинномозговой канал пациенту были получены из мозгового пузыря трех эмбрионов человека 12 недель гестации в лаборатории иммунологии ГУ НИИ акушерства, гинекологии и перинатологии РАМН (директор академик РАМН, профессор, доктор медицинских наук В. И. Кулаков, руководитель лаборатории иммунологии профессор Г. Т. Сухих). Весь материал предоставлялся данным академическим учреждением в госпиталь бесплатно, был стандартизирован и сертифицирован. То, что произошло с больным Кар-вым после интратекального введения клеточного препарата, можно было бы назвать только «чудом исцеления». Больной Кар-в, которому диагноз БАС был установлен ведущими неврологами страны в ГУ НИИ неврологии РАМН, с выраженным спастическим тетрапарезом, глубокими атрофиями мышц конечностей и прогрессирующими фибрилляциями мышц плечевого пояса и полной невозможностью к самостоятельному самообслуживанию, уже на пятый день после клеточной терапии отметил резкое уменьшение фибрилляций, к 14-му дню после трансфузии клеточного препарата у него стали уменьшаться атрофии мышц конечностей, и к 24-му дню после цитотрансфузии у него восстановилась функция самообслуживания: он стал сам одеваться, а к 30-му дню лечения стал сам застегивать большие пуговицы на больничной куртке. Через два месяца больной Кар-в был выписан из стационара и самостоятельно, на своих ногах, с опорой на трость, ушел из стационара. Данный случай наблюдали и обсуждали более 10 неврологов и нейрохирургов Центрального военно-морского клинического госпиталя. Мы сняли это «чудесное восстановление» на видеокамеру и демонстрировали всем желающим. После выписки из неврологического стационара у пациента Кар-ва продолжали уменьшаться атрофии мышц верхних и нижних конечностей в течение шести месяцев, значительно увеличился объем движений в них. К сожалению, мы не смогли увидеть дальнейшего «чуда полного излечения» пациента, так как через шесть месяцев после выписки он попал в автомобильную аварию и умер у себя на даче от жировой эмболии, полученной в результате переломов длинных трубчатых костей и полученных травм.
Последующие годы наша группа неоднократно пыталась повторить у других больных с БАС тот же результат нейровосстановления, как у больного Кар-ва, но, к сожалению, не удалось получить даже десятой части того успеха, полученного у самого первого пациента с применением препарата фетальных нервных клеток. Аналогичная ситуация характерна и для терапии клетками, полученными из пуповинной крови. И с 2003 г. мы полностью отказались от применения этих клеточных систем в лечении неврологических заболеваний. Если исследователь неспособен повторить собственный полученный результат, то эти исследования бесполезны и малонаучны. Я так и не смог повторить за все годы своего изучения клеточной терапии при БАС свой самый лучший результат. Однако мне стало очевидно, что в природе существует комбинация донорских клеточных и тканевых компонентов, которые способны реально восстановить поврежденные моторные нейроны и восстановить утраченные функции ГМ и СМ, даже несмотря на кажущуюся необратимость дегенеративно-атрофического процесса в нервной системе. Поэтому предложенная вам для ознакомления монография — это многолетняя попытка найти альтернативное решение в терапии БАС, используя стандартизированные клеточные продукты и лекарственные препараты, способные создать иммунные и гистотканевые предпосылки для восстановления нейроповреждений при БАС.
Мы построили эту книгу достаточно традиционно для монографической научной литературы. В первой главе книги проведен анализ актуальности изучения проблемы бокового амиотрофического склероза и даны современные определения понятия БАС и (или) БМН. В ней предпринята попытка представить последние исследования в области эпидемиологии БАС, поговорить об этиологии и патогенезе данного заболевания (глава вторая). Далее обсуждается спектр морфологических изменений при данном нейродегенеративном заболевании (глава третья). В четвертой главе монографии представлены основные известные протеомные изменения при боковом амиотрофическом склерозе. Пятая глава монографии посвящена фундаментальным аспектам избирательной дегенерации и проблеме эксайтотоксичности в биологии БАС, а также анализу повреждений гематоэнцефалического барьера при БАС на основе анализа концентрации нейроспецифических белков и антител к ним. Мы впервые публикуем эти данные, которые больше свидетельствуют об аутоиммунном генезе болезни, чем о первичной нейродегенерации. В шестой главе представлены существующие классификации БАС и систематизированы основные клинические проявления этого нейродегенеративного заболевания. Представлены собственные клинические наблюдения больных с БАС. Что касается седьмой главы книги, то в ней проанализированы основные информационно-коммутационные механизмы формирования ведущих клинических симптомов и синдромов при боковом амиотрофическом склерозе. Это достаточно специфичная глава, которая дает реальные представления о молекулярно-биологических механизмах формирования ведущих синдромов заболевания. Она дает новые представления для понимания и создания новой терапевтической стратегии молекулярно-нацеленного лечения БАС.
Глава восьмая поднимает проблему диагностики БАС, и в ней обсуждаются существующие диагностические алгоритмы выявления болезни. Абсолютно новый взгляд на иммунологию БАС нами сформулирован именно в девятой главе. Несомненно, что возможное решение проблемы эффективного лечения БАС может быть теоретически найдено, если будут отработаны молекулярно-биологические маркеры ранней диагностики этого заболевания. То есть диагностика заболевания должна будет осуществляться на самых ранних этапах болезни, когда большая часть мотонейронов ГМ и СМ еще сохранена. При этом показано, что ранняя молекулярно-биологическая диагностика БАС возможна и может быть реализована по анализу специфики изменений протеомного профиля поверхностных белков мембраны ГСК больного с БАС. Полагаем, что выявленные нами специфические маркеры белков клеточной поверхности ГСК, появляющиеся при БАС, являются нозоспецифичными для данного заболевания и диагностика БАС-специфичного профиля белков мембраны ГСК при БАС может стать важнейшим критерием ранней молекулярно-биологической диагностики этого заболевания. Насколько правильны наши доказательства и предположения в этой главе по ранней иммунодиагностике БАС, покажут время и дальнейшие фундаментальные исследования в данном направлении. Но наши находки проливают свет на механизмы развития данного заболевания, которые могут стать мишенями для целенаправленной терапии этого фатального заболевания.
В десятой главе представлены существующие взгляды ученых на классический конвенциональный подход к лечению БАС. Обсуждение его отчасти является формальным перечислением спектра современных лечебных, но малоэффективных мероприятий, которые сегодня активно используются в лечении БАС.
В одиннадцатой главе книги подробно обсуждаются экспериментальные модели БАС и способы применения клеточных препаратов на основе стволовых клеток у экспериментальных животных с моделями БАС. В этой главе даны обобщающие сведения авторов и научной литературы по клиническому трансляционному применению клеточных продуктов для терапии БАС у человека. В этой же главе книги представляются научные обобщения по достаточно новому направлению лечения БАС — методам нейромодуляции и нейрореабилитации. В тринадцатой главе этой книги представлена принципиально новая стратегия лечения больных с БАС, основанная на применении специальных клеточных и лекарственных препаратов и наших собственных научных изысканиях. Эта стратегия не является панацеей и пока не позволяет полностью излечить больного, но она дает возможность приостановить болезнь и статистически достоверно продлить время жизни пациента с БАС. В четырнадцатой главе книги представлены нейровосстановительная терапия и возможности реставрации нейродегенеративных нарушений мотонейронов с использованием современных биомедицинских клеточных продуктов. В настоящее время нет другого метода лечения БАС, позволяющего реально увеличить медиану времени жизни больных БАС на 30–40%.
В заключении монографии мы обобщили все наши научные находки и инновационные решения в диагностике и лечении БАС и сформулировали предполагаемые основные направления научных исследований БАС в ближайшем будущем.
В этом кратком вступлении мне хотелось бы поблагодарить сотрудников моей клиники и лично руководителя отделения нейрореабилитации клиники «НейроВита» врача-невролога Николая Ивановича Коваленко за помощь в курировании этого крайне тяжелого контингента неврологических больных и участие в научно-исследовательском изучении данного фатального заболевания, а также выразить признательность за терпимость и понимание среднего и младшего персонала клиники.
Автор будет рад принять все замечания и предложения по содержанию и клиническому смыслу изложенной в данной книге концепции ранней молекулярно-биологической диагностики и предложенной молекулярно-нацеленной (таргетной) стратегии лечения БАС по электронной почте neurovita-as@mail.ru.
Профессор, доктор медицинских наук
А. С. Брюховецкий
Глава 1. Актуальность проблемы, определения понятия и эпидемиология болезни
Впервые боковой амиотрофический склероз (БАС) был описан 150 лет назад физиологом Жан-Мартеном Шарко (J.-M. Charcot), которому удалось связать наблюдаемую у пациентов спастичность и патологию в спинном мозге (Rowland et al., 2001). Им было предложено название, которое используется до настоящего времени: слово «амиотрофический» отражает мышечную слабость и атрофию, а «латеральный склероз» обозначает склеротизацию передних и латеральных кортикоспинальных трактов, наблюдавшуюся им у больных с БАС (Wijesekera et al., 2009). В XIX в. в качестве самостоятельных клинических синдромов были описаны прогрессирующая мышечная атрофия (ПМА) (F. Aran, 1848), первичный боковой склероз (ПБС) (W. Erb, 1875) и прогрессирующий бульбарный паралич (ПБП) (A. Duchenne, 1860) (Бакулин и др., 2017). Так, Шарко рассматривал перечисленные состояния как отдельные синдромы, в то время как J. Dejerine и W. Gowers считали их проявлениями одной болезни. В статье «Проблема первичного бокового склероза», опубликованной в 1946 г., I.S. Wechsler и S. Brody отмечают, что клиника и течение ПБС, ПМА и БАС отличаются, что позволяет рассматривать их как отдельные дегенеративные состояния («склерозы») (Бакулин и др., 2017). Боковой амиотрофический склероз, или, сокращенно, БАС (англ. Amyotrophic Lateral Sclerosis, сокращенно — ALS, болезнь Шарко, болезнь Лу Герига, прогрессирующая мышечная атрофия), представляет собой фатальное заболевание двигательных нейронов, которое сегодня получило новое название — болезнь двигательных нейронов (БДН) (англ. motor neuron disease, MND). Заболевание характеризуется дегенеративными изменениями в верхних и нижних двигательных нейронах и приводит к прогрессирующей амиотрофии, фасцикуляциям, парезам, спастике, дыхательному параличу и смерти. Еще в 1933 г. W.R. Brain предложил использовать термин «болезнь двигательного нейрона» (БДН) для объединения клинически разных вариантов в одну общую диагностическую категорию. Сегодня этот термин в качестве названия заболевания является альтернативным названием БАС и достаточно прочно закрепился в современной зарубежной и отечественной клинической и научной неврологической литературе, хотя, по сути, не отражает всего диапазона известных к настоящему времени морфо-функциональных, геномно-постгеномных и информационно-коммутационных нарушений в нервной ткани, происходящих в организме пациента. Название БДН на самом деле больше отражает формальные морфологические и нейрофункциональные признаки болезни, чем ее сущность. Это название заболевания предполагает довольно примитивное понимание и представление о данной болезни как о локальном изолированном поражении первого и второго мотонейронов головного мозга (ГМ) и спинного мозга (СМ) человека. Мы считаем, что понимание патологии БАС исключительно как повреждения или заболевания моторных нейронов является фундаментальным научным заблуждением и очень ограничивает потенциальные возможности и перспективы для поиска реальной терапии. Другими словами, свести всю проблему возникновения БАС к проблеме только повреждения моторных нейронов теоретически и методологически неправильно и научно-практически неверно. Это, по сути, равносильно перестановке причины и следствия научного явления. Это понимание заболевания — не просто словесная эквилибристика, а скорее подмена понятий, и она является скорее научным тупиком, чем научным прорывом. Современное понимание БАС как болезни моторного нейрона — это проявление глобальных научных представлений устаревшего фундаментального системного анатомо-физиологического подхода к изучению нервных и психических болезней, который, к сожалению, не позволяет найти способа излечения от этого заболевания до настоящего времени. Если вы проанализируете все основные работы в области БАС за последнее время, то увидите, что новая формулировка заболевания как БМН привела к тому, что все исследователи сосредоточились на поиске внутренних, преимущественно генетических и эпигенетических причин этого заболевания, связанных непосредственно с повреждением мотонейронов, который пока не увенчался успехом. И скорее всего, это не даст нам ожидаемых результатов в будущем. Несомненно, в результате постоянных фундаментальных генетических исследований с секвенированием генома, изучением транскритомов экспрессии генов и протемного картирования моторных нервных клеток будут найдены новые, еще неизвестные патологические генетические, РНК- и протеомные патологические модификации в геноме, транскриптоме и протеоме мотонейронов, кроме уже известных 108 генетических мутаций в геноме и более 100 обнаруженных изменений белкового профиля мотонейронов у пациентов с БАС. Но это не даст нужного нам ответа и самое главное — не даст возможности найти нужное терапевтическое решение проблемы. Устаревший методологический инструментарий системного подхода в нейронауке, революционный в ХХ в., сегодня мешает пойти дальше, и понимание БАС как БДН является его классическим представлением и главным, по нашему мнению, «тормозом» учения о боковом амиотрофическом склерозе.
Повреждение моторных нейронов является исходом закономерной динамики формирования иммунных нарушений и дегенеративно-атрофического патологического процесса во всей нервно-мышечной системе пациента при БАС и следствием длительно текущей дегенерации и атрофии во всех клетках нервной ткани ГМ или СМ. Самым очевидным и демонстративным проявлением функционального и морфологического дефекта при данном заболевании является моторный дефект, но сведение всего спектра функциональных расстройств и патоморфологии к нему и определению болезни как исключительно болезни моторного нейрона ошибочно и резко ограничивает поиски средств для лечения этой страшной болезни. Нейротоксичность или эксайтотоксичность, в результате которой, как традиционно считается, идет собственно повреждение и дегенерация моторных нейронов при БАС, является только частью или важным звеном патогенетического процесса, но при этом почему-то не учитываются нарушения в других клетках нервной ткани, вовлеченных в патологический процесс в ГМ и СМ.
Но вернемся к существующим определениям понятия БАС. И. В. Моткова в своем диссертационном исследовании (2008) дает следующее определение БАС: боковой амиотрофический склероз — это нейродегенеративное заболевание, сопровождающееся гибелью центральных и периферических мотонейронов и характеризующееся прогрессирующим течением и летальным исходом. То есть в самом этом определении болезни заложена обреченность на фатальный исход болезни. Действительно, считается, что нервные клетки после дегенерации не имеют шансов на восстановление, и данное определение только констатировало факт наличия атрофического и дегенеративного процесса и его печальный исход. Сегодня работами большинства нейроученых по всему миру доказано, что нервные клетки способны регенерировать и восстанавливаться под воздействием различных типов стволовых клеток и факторов роста, поэтому летальность при БАС может быть преодолена. Однако именно очень поздняя диагностика БАС резко ограничивает возможности для подобного лечения и способствует увеличению количества больных с БАС.
Вот одно из наиболее характерных определений заболевания, найденное нами в интернете: БАС — это идиопатическое нейродегенеративное прогрессирующее заболевание неизвестной этиологии, обусловленное избирательным поражением периферических двигательных нейронов передних рогов спинного мозга и двигательных ядер ствола мозга, а также корковых (центральных) мотонейронов и боковых столбов спинного мозга. Ключевые слова в этом определении БАС — это «идиопатическое нейродегенеративное заболевание». Идиопатический (от греч. Idios — собственный, особый; pathos — страдание) означает «самостоятельный» (син. — эссенциальный). Идиопатической называют болезнь, которая возникает самостоятельно, то есть независимо от других поражений. При таких определениях болезни шансы на поиск лечения такого заболевания сокращаются до минимума.
В. И. Скворцова и Г. И. Левицкий (2004) дают свое клиническое определение этого заболевания: болезнь двигательного нейрона (БДН) — это нейродегенеративное заболевание, сопровождающееся гибелью центральных и периферических мотонейронов, неуклонным прогрессированием и летальным исходом. Клинически все строго и верно, но это определение не имеет будущего и ничего не объясняет для разработки и создания технологии лечения.
Д. Марфунин (2012) так определяет, что такое боковой амиотрофический склероз: БАС, или ALS, является фатальным, позднего начала, быстро прогрессирующим нейродегенеративным заболеванием, приводящим к мышечной слабости и атрофии, которые развиваются в паралич. Это, пожалуй, единственное определение, в котором автор определяет позднее начало заболевания как важнейший нозоспецифический фактор, и это важное качество этого определения.
С. А. Живолупов, Н. А. Рашидов, И. Н. Самарцев, С. А. Галицкий (2011) считают, что БАС является мультисистемным церебральным нейродегенеративным процессом. И. С. Бакулин с соавторами (2017) также считают, что боковой амиотрофический склероз (БАС) — это нейродегенеративное заболевание, протекающее в типичных случаях с поражением моторной коры, кортикоспинальных и кортиконуклеарных путей и периферических мотонейронов ствола и спинного мозга.
По современным данным, боковой амиотрофический склероз занимает третье место в структуре неврологических заболеваний и является одной из самых распространенных форм болезней двигательного нейрона (Honung et al., 2017). Распространенность БАС в разных странах неодинакова и в мире в среднем составляет от 0,8 до 7,3 случая на 100 000 человек в год, заболеваемость — 0,2–2,4 случая на 100 000 человек в год, при этом в последнее время отмечены тенденции к росту заболеваемости во всех возрастных группах и к более злокачественному течению (Abhinav et al., 2007; Julien, 2007; Karussis et al., 2010). Специалисты Национального института здоровья США подсчитали, что в США около 30 000 человек одномоментно болеют БАС, ежегодно диагностируется 5000 новых случаев, причиной смерти почти 300 000 ныне живущих американцев будет БАС, затраты на уход за одним пациентом в США составляют около 200 000 долларов в год (ALS CNTF Treatment Study Group, 1996). Последнее эпидемиологическое исследование БАС в СССР проводилось О. А. Хондкарианом и Г. А. Максудовым в 1970 г. и установило средний показатель заболеваемости 0,20,5/100 тыс. человек в год (Appel et al., 2008). В России на настоящий момент эпидемиологическая обстановка по БАС неизвестна, имеются лишь данные по отдельным городам и областям (Almer еt al., 2001; Марфунин, 2012). Проблема распространенности БАС в нашей стране требует дальнейшего изучения и уточнения. По последней статистике, ежегодно в мире выявляют 2–3 новых случая на 100 тысяч человек населения. Это небольшой процент встречаемости в общей частоте, но каждому пациенту необходимо оказывать специализированную помощь. Помощь необходима на всех этапах диагностики, а также после установки клинического диагноза (Висурханова и др., 2018). При всем при этом БАС представляет собой серьезную медицинскую и социальную проблему. У больных проявляются признаки и симптомы прогрессирующей мышечной атрофии и слабости, повышение утомляемости и проблемы с глотанием, результатом которых обычно становятся респираторная дисфункция и гибель (Rowland, 1994; Sejvar, 2005). Прогрессирующий неврологический дефицит функциональности приводит к общей утрате самостоятельности (Ng, 2011), полной трудоспособности и невозможности самообслуживания. Медиана выживаемости больных с БАС в зависимости от формы заболевания составляет 2–6 лет с момента постановки диагноза. Исход БАС, как правило, мучительный и трагичный. Больной умирает в полном сознании из-за нейрогенной остановки дыхания. Попытки противостоять такому исходу заболевания приводят к тому, что родственники отдельных больных переводят их на искусственную вентиляцию легких и продляют их жизнь на годы. В моей практике один из больных прожил на искусственной вентиляции легких 7 лет, другая больная из Болгарии прожила на аппаратном дыхании 12 лет и жива по настоящее время. Однако качество жизни у полностью обездвиженных больных БАС на аппаратном дыхании оставляет желать лучшего. Особенностью этих больных является почти полное сохранение интеллектуально-мнестических функций мозга и продуктивной интеллектуальной деятельности, как правило, до летального исхода.
БАС поражает лиц преимущественного зрелого и трудоспособного возраста (20–80 лет), с высоким интеллектуальным и профессиональным потенциалом, неизбежно приводит к тяжелой инвалидности и смерти больных, что в сочетании с отсутствием эффективных методов лечения обосновывает актуальность изучения этой болезни. Прогредиентность течения заболевания, быстрая смена легких двигательных расстройств тяжелыми парезами и параличами делают проблему ранней диагностики заболевания первостепенной, прежде всего для практического здравоохранения. Большой интерес в этом плане представляет группа клинически сходных с БАС нозологий, требующих дифференциальной диагностики и принципиально иных, потенциально эффективных подходов в лечении. Кроме того, единственный препарат, показавший свою эффективность в отношении БАС, рилузол, должен назначаться пациентам с установленным диагнозом как можно раньше. Поэтому необходимы критерии, на основании которых можно с более высокой достоверностью судить о наличии у пациента БАС или синдрома БАС в его дебюте и (или) при первом осмотре.
Заболеваемость спорадическим БАС в 1990-х гг. составляла от 1,5 до 2,7 на 100 000 населения в год (средний показатель составлял 1,89 на 100 000 в год) в странах Европы и США (Worms, 2001). Частота вновь выявленных заболеваний при осмотре населения в определенный момент времени варьируется в диапазоне от 2,7 до 7,4 случая на 100 000 (средняя величина составляет 5,2 на 100 000) в странах Запада (Worms, 2001). Риски возникновения спорадического БАС к 70-летнему возрасту оцениваются в 1 случай на 1000 (Traynor et al., 1999), но более точные оценки, скорее всего, составляют 1 случай на 400 (Johnston et al., 2006). Непротиворечивые данные, полученные в результате проведения исследований, говорят о небольшом превышении количества мужчин, подверженных возникновению заболевания, над количеством женщин, при коэффициенте соотношения мужчин к женщинам, равном 1,5/1. Более свежие данные дают основания предположить, что гендерное соотношение может приближаться к равным значениям (Worms, 2001; Abhinav et al., 2008; Logroscino et al., 2008). Уровень смертности в результате БАС в 1990 г. варьировался от 1,54 до 2,55 на 100 000 в год, а проведенное в более недавнее время исследование свидетельствует о 1,84 смерти на 100 000 населения (Worms, 2001; Sejvar et al., 2005). Средний возраст начала развития случаев спорадического БАС варьируется между 55 и 65 годами, где средний возраст возникновения заболевания составляет 64 года (Haverkamp et al., 1995). Только в 5% случаев наступление заболевания выявлялось в возрасте моложе 30 лет, хотя случаи ювенального спорадического БАС регистрируются все чаще (Gouveia, de Carvalho, 2007). Бульбарные проявления заболевания более распространены среди женщин и в группах старшего возраста, где 43% больных старше 70 лет демонстрируют бульбарные симптомы в сравнении с 15% больных моложе 30 лет (Beghi et al., 2007; Forbes et al., 2004).
Глава 2. Этиология и патогенез заболевания
Несмотря на то что заболевание описано более 150 лет назад, его этиология и патогенез остаются не до конца изученными, а эффективные методы лечения отсутствуют в принципе. Это связано с тем, что мы пока не понимаем истинных причин и патогенетических механизмов возникновения и динамики этой болезни. Существует огромное количество теорий возникновения БАС, что лишь свидетельствует об отсутствии единственно правильной теории развития этой нейродегенеративной болезни. Одной из первых теорий возникновения БАС была вирусная теория, в котоhой анализировалась роль энтеровирусов, вируса ВИЧ и прионов в возникновении БАС. Другие исследователи пытались доказать участие вируса полиомиелита в формировании болезни. Вирусная теория была популярна в 60–70-х гг. ХХ в., но так и не нашла своего подтверждения. Ученые США и СССР проводили опыты на обезьянах, вводя им экстракты спинного мозга больных людей, но эффекта возникновения БАС отмечено не было. Применение современных противовирусных препаратов оказалось малоэффективным в лечении БАС, в связи с чем доказательность вирусной природы болезни была также опровергнута, так как новые противовирусные средства были эффективны против основных нейровирусов, диагностируемых у человека, как in vitro, так и in vivo (Левицкий, 2003; Скворцова, Левицкий, 2004).
Долгое время считалось, что в результате вирусной нейроинфекции (корь, краснуха, герпес, цитомегаловирус и т.д.) у пациента в нервной ткани головного и спинного мозга возникает аутоиммунный процесс, как и при рассеянном склерозе. При этом считалось, что аутоиммунный процесс при БАС запускался после повреждения боковых столбов спинного мозга (СМ) нейроинфекцией. В современной классической неврологии ХХ в. БАС также относили к группе аутоиммунных заболеваний и, как правило, описывали как одну из специфичных разновидностей аутоиммунного процесса в нервной системе, наряду с рассеянным склерозом и рассеянным энцефаломиелитом. Аутоиммунная теория возникновения БАС и сегодня не утратила своей актуальности. Эта точка зрения на причины развития БАС основывалась на существующем научном представлении об инфекционно-вирусном и воспалительном генезе заболевания, которые и запускают аутоиммунный процесс, в результате которого образуются аутоиммунные антитела. Но на самом деле до настоящего времени истинная природа БАС не ясна. Более того, по мнению целого ряда отечественных исследователей, аргументов в пользу аутоиммунного процесса в мозге при этом заболевании недостаточно. Основными аргументами, которые современные неврологи приводят против аутоиммунной теории БАС, является низкая терапевтическая эффективность глюкокортикоидных гормонов, неэффективность плазмофереза, низкая эффективность иммуноглобулинов и цитостатиков, неэффективность аутологичной трансплантации костного мозга (ТКМ) и неэффективность блокаторов цитокиновых рецепторов (Левицкий, 2003; Скворцова, Левицкий, 2004; Скворцова и др., 2005). Ведущие отечественные неврологи в области БАС (Скворцова и др, 2005; Скворцова, Левицкий, 2007) считают, что аутоиммунная теория не получила подтверждения в большинстве современных исследований, что аутоиммунные нарушения при БДН носят вторичный характер. На поздних стадиях болезни у пациентов может развиться вторичный иммунодефицит на фоне дисфагии и алиментарной недостаточности (Левицкий, 2003). Мы считаем (Брюховецкий, Хотимченко, 2018), что подобная точка зрения, основанная на неэффективности лекарств и методов лечения при аутоиммунном характере заболевания, правомерна, но может быть применима только в том случае, если классический аутоиммунный процесс протекает по воспалительному (экссудативному) типу и в его структуре работают эффекторные механизмы иммунокомпетентных клеток. В этом случае действительно против воспаления будут работать глюкокортикоидные гормоны, будет эффективен плазмоферез и элиминация антител и т. д. Но ведь аутоиммунный процесс может идти по пролиферативному или фиброзирующему типу, и в этом случае в генезе болезни будут задействованы эпигенетические механизмы иммунокомпетентных клеток. Двойственность аутоиммунного процесса, с одной стороны, проявляется тем, что существуют определенные стадии аутоиммунного процесса: 1) гранулематозно-воспалительная (острая), 2) диффузно-пролиферативная (подострая) и 3) фиброзная (хроническая). И тогда очевидно, что острый и подострый процесс пойдут по типу воспаления, а хронический аутоиммунный процесс при БАС обязательно пойдет по типу фиброзного процесса и может проявиться нейродегенерацией мотонейронов. С другой стороны, если в развитии болезни задействованы только периферические иммунокомпетентные клетки, то аутоиммунный процесс обязательно будет острым или подострым, по типу воспаления. Но если в генезе заболевания задействованы патологические гемопоэтические стволовые и прогениторные клеточные гемопоэтические системы, то все потомки этих клеточных систем вызовут пролиферативный (фиброзирующий) процесс, и тогда эффективность лекарственных средств, эффективных в коррекции воспаления, станет минимальной, что и возможно при БАС. Поэтому аутоиммунный генез этой болезни исключать нельзя, и мы к этому вернемся ниже.
Другая точка зрения на этиологию и патогенез развития БАС — сосудистая теория возникновения данного заболевания. В основе этой теории лежали существующие научные представления об обязательном сосудистом компоненте как фундаментальной причине возникновения БАС у человека. Сосудистый фактор в развитии БАС всегда считался и считается одним из важнейших в патогенезе заболевания. Роль сосудистых нарушений при БАС отмечали почти все классические неврологи русской школы (Карлов, 1987, 2002; Акимов, 1992). Важное значение сосудистого фактора в развитии БАС связано с анатомическими особенностями кровоснабжения боковых столбов спинного мозга, где расположено основное место повреждения СМ и где теоретически мог бы разворачиваться весь сценарий патологических нарушений. Особенностью кровоснабжения спинного мозга в целом и боковых столбов спинного мозга в частности является вхождение кровеносных питающих сосудов в проекции боковых столбов СМ каждого сегмента спинного мозга как основной питающей артерии спинного мозга (радикуло-медулярная артерия), которая на уровне шейного утолщения СМ определяется как передняя радикуло-медулярная артерия. В грудном отделе она представлена как большая передняя радикуло-медулярная артерия (а. Адамкевича), а на уровне последних сегментов спинного мозга она называется нижней дополнительной радикуло-медулярной артерией (а. Депрож-Готтерона). Эти артерии берут свое начало в межреберных артериях. Все эти артерии на всем протяжении СМ соединяет передняя спинальная артерия, которая является основным ответвлением передней спинальной артерии (а. spinalis ant.), входящей в СМ. Сзади мозг питают задние спинальные артерии. Таким образом, боковые столбы спинного мозга, наиболее сильно страдающие при БАС, первыми получают кровоснабжение в СМ, и резонно было бы предположить, что даже самые незначительные микронарушения гемостаза и кровоснабжения СМ могут сказаться на функции боковых столбов СМ, где, собственно, и расположены моторные нейроны. Ряд исследователей (Брюховецкий и др., 1996; Зубрицкий и др., 1998) предполагали возможность нарушения микроциркуляции в зоне кровоснабжения боковых столбов спинного мозга при БАС и исследовали состояние сосудов спинного мозга с использованием спинальной и церебральной ангиографии у 15 больных с БАС. Было установлено, что почти у всех больных имелся определенный вегетативный дефект иннервации церебральных и спинальных сосудов или их аномальное строение: у 8 больных были выявлены явления s-образной патологической извитости общей сонной или внутренней артерии, гипоплазия (аплазия) правой или левой позвоночной артерии у 5 пациентов, или комбинация этих признаков у 4 больных. У 2 пациентов была выявлена патологическая извитость правой позвоночной артерии, а у одного пациента с шейной формой БАС — патология развития сосудов ГМ (незамкнутость) Виллизиева круга. Было высказано предположение, что в генезе БАС сосудистые расстройства являются важным пусковым механизмом развития болезни. При травматическом, вирусном или воспалительном повреждении сосудов, кровоснабжающих СМ, токсическом воздействии на них или воздействии других этиологических факторов БАС в первую очередь формируется парез или паралич нервов автономной нервной системы, иннервирующих эти сосуды. Это приводит к нарушению вегетативного обеспечения этих сосудов, что и было зарегистрировано при церебральной или спинальной ангиографии в форме образования патологических извитостей. Вследствие этих этиопатогенетических изменений формируется кинкинг, или «провисание», участка паретичного сосуда. Появление паретичной части магистрального сосуда головы или сосуда СМ приводит к замедлению циркуляции крови в этой зоне и формированию микротромбов и микроагрегаций форменных элементов крови. Микротромбы, возникающие в крупных сосудах в зоне кровоснабжения СМ, «забивают» более мелкие сосуды, расположенные именно в зоне вхождения спинальных артерий в СМ и нарушают кровоснабжение в зоне боковых столбов СМ. Мы полагаем, что микрососудистые нарушения при БАС являются основным триггером комплекса или каскада необратимых биохимических реакций во всех клетках СМ. Косвенным фактором правомерности этого предположения были временные позитивные клинические результаты в лечении БАС при региональной перфузии спазмолитических средств (но-шпа, папаверин и т.д.) рентгенохирургически непосредственно в спинальную артерию зоны дегенерации и повреждения СМ. Мой личный последующий опыт выявления патологических извитостей магистральных сосудов шеи (преимущественно внутренней сонной артерии на стороне клинических проявлений) у всех обследуемых пациентов с БАС показал, что в 65% случаев действительно у больных БАС удается выявить патологические извитости (кинкинги) магистральных артерий, кровоснабжающих спинной мозг. Появление рентгенохирургических техник и возможность проведения малоинвазивных внутриартериальных исследований в конце 1990-х и начале 2000-х гг. позволило установить наличие реальных патологических извитостей магистральных артерий, кровоснабжающих спинной мозг, у пациентов с БАС. Однако попытки проведения рентгенохирургической программной региональной перфузии этих зон спинного мозга вазоактивными и тромболитическими препаратами не увенчались успехом и не приводили к клиническому успеху. Из 20 пациентов с БАС, имеющих патологические извитости магистральных артерий, только у 5 пациентов было отмечено незначительное и временное смягчение клинических проявлений заболевания. По-видимому, формирование патологических извитостей магистральных артерий, кровоснабжающих дегенеративные участки головного и спинного мозга, при БАС является следствием системной дегенерации тканевого модуля нервной ткани. Наиболее вероятно, появление патологических извитостей магистральных артерий, кровоснабжающих дегенеративные участки спинного мозга, является следствием, а не причиной заболевания.
Д. Марфунин (2012) в своей исследовательской работе «О боковом амиотрофическом склерозе» обсуждает варианты сочетаний вирусного начала и сосудистых нарушений при БАС. Он абсолютно верно утверждает, что верхние моторные нейроны расположены в двигательной области коры головного мозга, то есть в прецентральной извилине. Мышцы тела имеют определенную проекцию на этой извилине. Известно также, что эта извилина получает артериальную кровь из двух артерий — передней мозговой и средней мозговой. Граница зон кровоснабжения передней и средней артерии лежит приблизительно между проекцией тела и головы. Как считает Д. Марфунин, соблазнительно предположить, что поражающий агент (если такой существует) мог проникнуть в извилину извне. Учитывая вышеописанную картину прогрессии болезни, можно предполагать, что этот агент должен быть живым, способным к самовоспроизводству и мог бы лежать в основе и быть причиной болезни. Но попытки обнаружить вирусные частицы и молекулы в пределах моторных нейронов, корковых или спинальных, или в пределах тканей скелетных мышц были безуспешными. Более того, при рассмотрении пула моторных нейронов пациентов БАС в пределах пораженной области обнаруживается лишь недостаточность нейронов, но нет никакого намека на какую-либо борьбу нейрона против предполагаемого внедрившегося агента, способного его разрушить, только умеренная воспалительная реакция в окружающих тканях, главным образом основанная на наличии реактивной микроглии. Если все же предположить, что такой агент существует, то возникает несколько вопросов. Во-первых, кто он? Во-вторых, почему поражает лишь моторные нейроны? Почему поражает весь моторный тракт от терминальной пластинки до верхних моторных нейронов? Почему этот тракт поражается не одновременно, а начинается с терминальной пластинки и далее процесс распространяется вверх, создавая картину «отмирания»? И, наконец, почему моторные нейроны, не сопротивляясь, запускают процесс апоптоза? Если в качестве ответа на один из этих вопросов предположить, забегая вперед, что моторный тракт поражается с целью (если таковая существует) достижения гипокинезии, то для этого достаточно разрушить терминальную пластинку или, в крайнем случае, аксон переднего рога. Зачем же поражается весь тракт, вплоть до клеток Беца? Реального ответа нет. Ни вирусная, ни сосудистая теории не дают ответа на эти вопросы.
Другую причину возникновения БАС предлагает посттравматическая теория. Изучив анамнез более 60 пациентов с БАС, а также 20 пациентов с БАС, имеющих объективно доказанные патологические извитости магистральных артерий шеи и артерий, кровоснабжающих спинной мозг, мы обратили внимание на наличие в анамнезе этих больных серьезной закрытой травмы позвоночника или головного мозга. Этот факт подтвердился почти у половины этих больных. В трех случаях возникновение болезни отмечалось через 6–8 месяцев после минно-взрывных повреждений (боевая травма, криминальная травма при взрыве в автомобиле и т.д.), в 10 случаях удавалось выявить тяжелые повреждения позвоночника при профессиональной спортивной травме или занятиях экстремальными видами спорта: горные лыжи, единоборства, падение с высоты, падение на мотоцикле, неудачное падение спиной при прыжках с парашютом и т. д. Однако убедительных подтверждений этого феномена мы не отметили, моделирование этого феномена на экспериментальных животных не приводило к развитию клинической картины болезни БАС (Брюховецкий и др., 1998). Возможно, модель травмы спинного мозга на крысах не может служить объективной моделью хронического сосудистого фактора в возникновении БАС у человека из-за серьезных отличий анатомии сосудов и особенностей, связанных с прямохождением человека. Необходимо смоделировать эту нозологию на обезьянах и окончательно отказаться от этой теории или доказать ее состоятельность.
Существует и другая точка зрения, объясняющая возникновение БАС у спортсменов. Это теория чрезмерной физической активности. Согласно этой гипотезе, риск заболеть БАС у профессиональных футболистов в 6,5 раза выше, чем у обычного населения (Chio et al., 2005). A. Al-Chalabi, P.N. Leigh (2005) показали, что травмы, полученные при игре в футбол, являются причиной возникновения БАС у целого ряда профессиональных игроков. Напомним, что в США болезнь впервые была описана у игрока в американский футбол Лу Герига (болезнь Герига). Это связано с рядом специфических факторов: 1) с физической активностью, независимо от вида спорта; 2) с микротравмами или спецификой физических упражнений; 3) с употреблением допинга; 4) с типичными для футбола экологическими факторами; 5) с генетическими факторами, которые связаны с чрезмерной физической работоспособностью.
Есть научно обоснованная гипотеза возникновения БАС как паранеопластического процесса. Описаны случаи сочетания БАС с раком легкого, раком молочной железы, раком щитовидной железы, раком кишечника, инсулиномой и даже с мультиформной глиобластомой. Однако в последующем было доказано, что во всех этих случаях БАС был самостоятельным заболеванием. Наши собственные исследования состояния иммунной системы при различных типах рака и при БАС показали, что в основе этих заболеваний лежит хроническая или острая иммунная недостаточность, которая обусловлена разноуровневыми и разнонаправленными мультимаркерными изменениями протеомного профиля клеточной поверхности гемопоэтических стволовых клеток (ГСК) пациента. Учитывая, что ГСК у каждого человека являются основным источником образования всех типов клеток его иммунной системы, глобальные отличия протеомного профиля этих клеток между собой позволяют утверждать, что эти заболевания имеют различные нозоспецифичные иммунные нарушения. Профиль маркеров белков клеточной поверхности ГСК при раке, у больных с БАС и у здоровых доноров костного мозга имеют фундаментальные отличия между собой, и они были картированы и профилированы нами и представлены в главе 8 этой книги.
Существует также теория влияния экзогенных факторов на возникновение болезни. Экзогенными этиологическими факторами являются специфические воздействия окружающей природы. БАС даже получил название «болезнь Гуам», в которой сочетается клиника бокового амиотрофического склероза (БАС), паркинсонизма и деменции (комплекс Гуам) — редкая эндемическая патология, в основе которой лежит прогрессирующая генерализованная дегенерация нейронов центральной нервной системы (Morris et al., 2004; Скворцова и др., 2005; Яхно, 2005; Завалишин, 2009). Впервые это заболевание было описано D.R. Koerner в 1952 г. у коренных жителей чаморро на острове Гуам в Марианском архипелаге, расположенном в восточной части Тихоокеанского бассейна. Были подробно изучены воздействия ряда токсинов в природной экосистеме на острове Гуам. По данным P.A. Cox et al. (Сox et al., 2003), S.J. Murch et al. (Мurch et al., 2004), одним из природных небелковых веществ, обладающих нейротоксическим действием, является beta-Methylamino-L-alanin. Это вещество синтезируют цианобактерии, расположенные на кораллах, которыми, в свою очередь, питаются морские черепахи. Частое употребление местными жителями мяса черепах приводит к повышению его концентрации в тканях в 10–240 раз. Таким образом, в пищевой цепи на острове Гуам формируется эндогенный нейротоксический резервуар, оказывающий влияние на метаболизм белков, что способствует развитию заболевания. По мнению S. Murch et al., этот механизм приводит к развитию болезни даже через несколько лет у чаморро, покинувших Гуам (Мurch et al., 2004).
D.R. Koerner (1954) впервые обратил внимание на необычное сочетание неврологических синдромов, частота которых в 50–100 раз превышала распространенность бокового амиотрофического склероза в других странах мира, который нередко носил семейный характер (Koerner, 1952). Позднее эта патология была описана в этом же регионе на полуострове Кии в Японии и в западной части Новой Гвинеи (Hermosura et al., 2009; Kokubo, Kuzuhara, 2003). Заболевание чаще встречается у мужчин (соотношение муж.: жен. — 1,7:1) в широком возрастном диапазоне — от 30 и свыше 70 лет (Morris et al., 2001). Максимальная частота приходится на возраст 55–64 года. Болезнь чаще имеет быстрый и фатальный характер. В качестве спорадических случаев БАС и фронтотемпоральная деменция описаны в других странах мира (Егоркина, Гапонов, 2007; Dickson, 2008; Hasegawa et al., 2007). Патогенез болезни Гуам связан с формированием многочисленных нейрофибриллярных включений, преобладающих в коре, базальных ганглиях, гиппокампе, миндалевидном теле, спинном мозге, которые приводят к дегенерации соответствующих нейронов. Причиной конформационных нарушений в нейронах считают изменения в гене, кодирующем синтез микротубулярного тау-протеина (Morris et al., 2001; Hasegawa et al., 2007). Наrry Zimmerman (2001) показал, что заболеваемость БАС у коренного населения острова Гуам, расположенного в Тихом океане, составляла около 70 на 100 000 населения и была связана с питанием черепаховым мясом, которое использовали аборигены в пищу. При ограничении этой еды в пищу заболеваемость БАС на Гуам снизилась к концу века с 70 до 7 на 100 000 населения. Этот феномен получил название «комплекс острова Гуам».
Несмотря на то что удалось идентифицировать некоторые генетические факторы риска, фундаментальные причины, лежащие в основе БАС, продолжают оставаться неясными. Недавний пересмотр факторов риска внешней (окружающей) среды, выступающих в качестве этиологии БАС, не позволил прийти к установлению последовательной взаимосвязи между отдельным фактором внешней среды и риском развития болезни. За последнее десятилетие в области изучения этиологии и патогенеза БАС достигнуты определенные успехи.
Одной из красивых гипотез возникновения БАС является нейрофиламентная теория. Ее смысл заключается в том, что моторные нейронные клетки это самые большие нейроны в человеческом организме. Сохранение структуры нейрона и транспортные процессы в нем обеспечиваются цитосклелетом этой клетки. Белки цитоскелета — это в первую очередь нейрофиламенты — микроскопические системы трубочек и каркаса клетки. Мутантная СОД-1 может взаимодействовать с нейрофиламентами, что нарушает аксональный транспорт.
Также одним из малоизученных аспектов патофизиологии БАС является функциональное состояние сегментарного аппарата спинного мозга и внутри- и надсегментарных влияний на периферический мотонейрон. Этот аспект патогенеза БАС представляется актуальным для дальнейшего исследования, поскольку изучение изменений на уровне сегмента спинного мозга может дать возможность обоснования новых методов лечения с целью оптимальной фармакологической коррекции афферентно-эфферентной дисфункции сегментарного уровня. Эта теория получила название гипотезы восходящей и нисходящей нейродегенерации. БАС традиционно рассматривается как заболевание двигательной системы. Однако существуют работы, подтверждающие мультисистемный характер БАС с поражением экстрамоторных отделов центральной нервной системы (ЦНС) (Appel et al., 2008; Ordes et al., 2011; De Vos et al., 2008; Meininger et al., 2000) и экстранейральных систем (Meininger et al., 2004). Когнитивные нарушения, отражающие экстрамоторное поражение мозга, долгое время рассматривались как исключительные клинические варианты случайного развития деменции у 3–5% пациентов с БАС (Beauverd et al., 2012). Но в настоящее время все больше подтверждений находит гипотеза, что БАС и фронтотемпоральная деменция (ФТД) имеют общие компоненты патогенеза или даже являются различными проявлениями одного заболевания. Относительно высокая частота деменций при БАС подчеркивает необходимость разработки методик для краткого нейропсихологического обследования пациентов. Кроме уточнения патогенетических моментов и коррекции ведения пациента с учетом наличия у него когнитивных расстройств и угрозы развития деменции, ментальная сфера пациентов с БАС может стать предметом тщательного обследования, если встает юридический вопрос о дееспособности пациента. Большинство авторов выступают в поддержку сложносоставных взаимодействий на уровне генетической микросреды, выступающих в качестве предполагаемого причинного фактора дегенерации двигательных нейронов (Shaw, 2005; Cozzolino et al., 2008). Были исследованы гипотетические экзогенные факторы риска БАС, и их список может быть представлен следующими экзогенными факторами риска развития спорадических форм БАС: пищевые (диетические) факторы, электрическая травма, семейный анамнез болезни Альцгеймера или Паркинсона, географическое место жительства, служба в армии, детородный возраст, количество родов (у женщин) и порядок родов, возраст при наступлении менопаузы, потеря ребенка, род занятий, физическая активность, игра в футбол на профессиональном уровне, предыдущие полиомиелитные инфекции, раса и этническая принадлежность, курение, воздействие токсинов (сельскохозяйственные химические вещества, свинец), травмы позвоночника, стаж образования.
Генетические факторы. Примерно у 10% людей с заболеванием БАС имеется по крайней мере еще один член семьи, подвергшийся воздействию данной болезни, и предполагается, что в анамнезе может быть обнаружен семейный след развития БАС. Около 20% больных с аутосомной доминантной формой семейного БАС (СемБАС) и 2% больных с формой спорадического БАС (СпорБАС) обнаруживают мутации гена супероксиддисмутазы меди цинка SOD1 (Rosen et al., 1993). Предполагается, что мутации гена SOD1 вызывают развитие заболевания посредством токсического приобретения функции, а не через механизм утраты антиоксидантной функции фермента SOD1 (Shaw, 2005). К другим генам, вызывающим развитие семейной формы БАС (семБАС), можно отнести: алсин (alsin, БАС2); сенатаксин (senataxin, БАС4 и БАС5); везикуло-ассоциированный мембранный протеин (VAPB, БАС8), ангиогенин (angiogenin) и мутацию, происходящую в подгруппе p150 динактина (dynactin) DCTN1; оптинеурин (optineurin, ALS12). В недавнее время у пациентов с семейной и спорадической формами БАС (семБАС, спорБАС) были обнаружены мутации гена TARDBP, кодирующего TAR ДНК-связывающий протеин 43 (TDP-43), расположенный в хромосоме 1p36.22. Более того, мутации гена fused in sarcoma (FUS) недавно были идентифицированы примерно у 4% больных с семейной формой БАС, негативной к гену SOD1. Некоторые мутации других видов генов, способные увеличивать восприимчивость к БАС, определялись и у спорадических пациентов, в частности, мутации в KSP повторной области гена тяжелого нейрофиламента (NEFH), кодирующего нейрофиламентные тяжелые производные единицы; мутации в генотипе аполипопротеина Е Σ4 (apolipoprotein E Σ4 genotype); мутации в сниженной экспрессии протеина транспортера 2 возбудительной аминокислоты EAAT2. Сообщалось также о связи трех мутаций VEGF гена с повышенным риском развития спорадического БАС (спорадических случаев заболевания), в то время как недавно проведенный многоплановый метаанализ не выявил взаимосвязи между гаплотипами гена VEGF и увеличением риска возникновения БАС у людей. Подобные вариационные изменения отмечались в генах-кандидатах или при выполнении общегеномных исследований ассоциативных связей (Dion et al., 2009).
Курение как патогенетический фактор БАС. Методы доказательной медицины подтвердили связь курения с развитием БАС (Chen Lien al., 2015).
На сегодня не существует общепризнанной гипотезы патогенеза бокового амиотрофического склероза. Согласно современным представлениям, развитие БАС обусловлено взаимодействием наследственных и экзогенных провоцирующих факторов. Множество патологических изменений в нейронах приводит к предположению о многовариантном этиологическом факторе.
Окислительный стресс считается важнейшим фактором в этиопатогенезе БАС. Известно, что окислительный стресс причастен к процессу нейродегенерации при БАС. Также известно, что скопление образцов активных форм кислорода вызывает гибель клеток. Поскольку мутации, происходящие в гене SOD1, могут привести к семейному типу БАС, этот механизм окислительного стресса дегенерации двигательных нейронов при БАС представляет научный интерес. Данная гипотеза поддерживается биохимическими изменениями, отражающими повреждения свободных радикалов и патологический метаболизм свободных радикалов в цереброспинальной жидкости и образцах аутопсий пациентов с БАС (Ferrante et al., 1997; Smith et al., 1998; Tohogi et al., 1999). Кроме того, у образцов фибробластов, культивированных у больных с БАС, выявлялась повышенная чувствительность к окислительным разрушениям по сравнению с участниками контрольных групп (Aguirre et al., 1998).
Предполагается, что перекись водорода может служить аномальным субстратом для конформированной молекулы SOD1. В результате происходит усиление пероксидантных реакций и возрастает производство токсичных гидроксильных радикалов. Существенная роль окислительного стресса в патогенезе БАС подтверждается биохимическими исследованиями, при которых у больных обнаружилась недостаточность ряда систем антиоксидантной защиты, дисфункции митохондрий, дисметаболизм глутатиона, эксайтотоксина глутамата и механизмы глутаматного транспорта. Возможно, окислительное повреждение белковых мишеней (SOD1, нейрофиламентных белков, альфа-синукленина и т.д.) может облегчать и ускорять их совместную агрегацию, формирование цитоплазматических включений, которые служат субстратом для дальнейших патохимических окислительных реакций.
Апоптоз — опосредованная каспазами гибель клеток. Предполагается, что окончательный процесс гибели двигательных нейронов при БАС напоминает апоптозный путь запрограммированной клеточной гибели. Апоптоз представляет собой механизм, посредством которого двигательные нейроны отмирают в моделях мутантных мышей с трансгенным SOD1. В этих моделях были последовательно активированы апоптозные сигналы каспазы-1 и -3. Хроническая активация каспазы-1 происходила в раннем пресимптоматическом периоде, а каспаза-3 была активирована намного позже, будучи финальным эффектором гибели клеток (Pasinelli, 2000). В соответствии с данными сообщениями, внутрицеребровентрикулярная доставка широкого ингибитора каспазы приводила к снижению mRNA уровней каспазы-1 и -3 в тканях спинного мозга мутантных мышей с трансгенным SOD1 (G93A). Кроме того, двигательные нейроны были сохранены у мышей, прошедших трансфузию ингибитором каспазы, в сравнении с мышами, получившими трансфузию носителя-растворителя. Также удалось задержать начало развития заболевания и его прогрессирование (Li et al., 2000). Биохимические маркеры апоптоза определяются в терминальной стадии БАС пациентов и животных моделей. Ключевые элементы пути здорового апоптоза вовлечены в гибель клеток при БАС, в том числе семейство каспаз протеолитических ферментов, Bcl2 семейство онкопротеинов (антиапоптозные и проапоптозные онкогены) и ингибирующее апоптоз семейство протеинов. Что касается дополнительного подтверждения, говорящего в пользу апоптозного механизма гибели нейронов, было замечено, что избыточная экспрессия антиапоптозного протеина Bcl-2 (Kostic et al., 1997) и удаление проапоптозного протеина Bax (Gould et al., 2006) способны удерживать и сохранять двигательную функцию и продлевать выживаемость мутантных мышей с трансгенным SOD1 (G93A). Таким образом, токсичность мутантного SOD1, по всей видимости, опосредуется, по крайней мере частично, каспазами и другими апоптозными факторами.
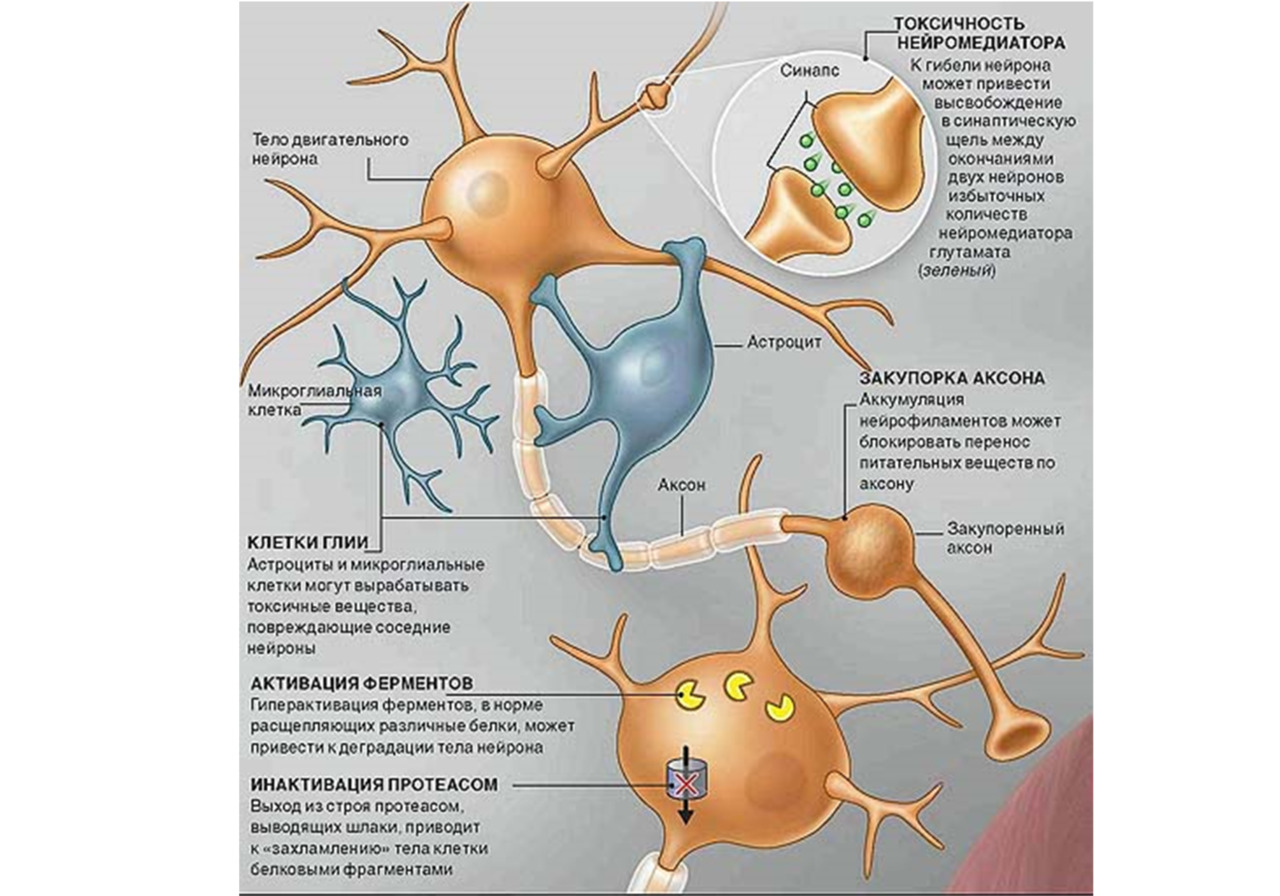
Рис. 1. Установленные негенетические молекулярно-биологические механизмы развития бокового амиотрофического склероза
В интернете мы нашли очень познавательную схему патофизиологических нарушений и молекулярно-биологических механизмов, происходящих в нервной ткани при БАС, и приводим ее для иллюстрации всего вышесказанного (рис. 1).
Представленная схема позволяет очень наглядно увидеть все основные молекулярно-биологические механизмы патогенеза данного заболевания (рис. 2).
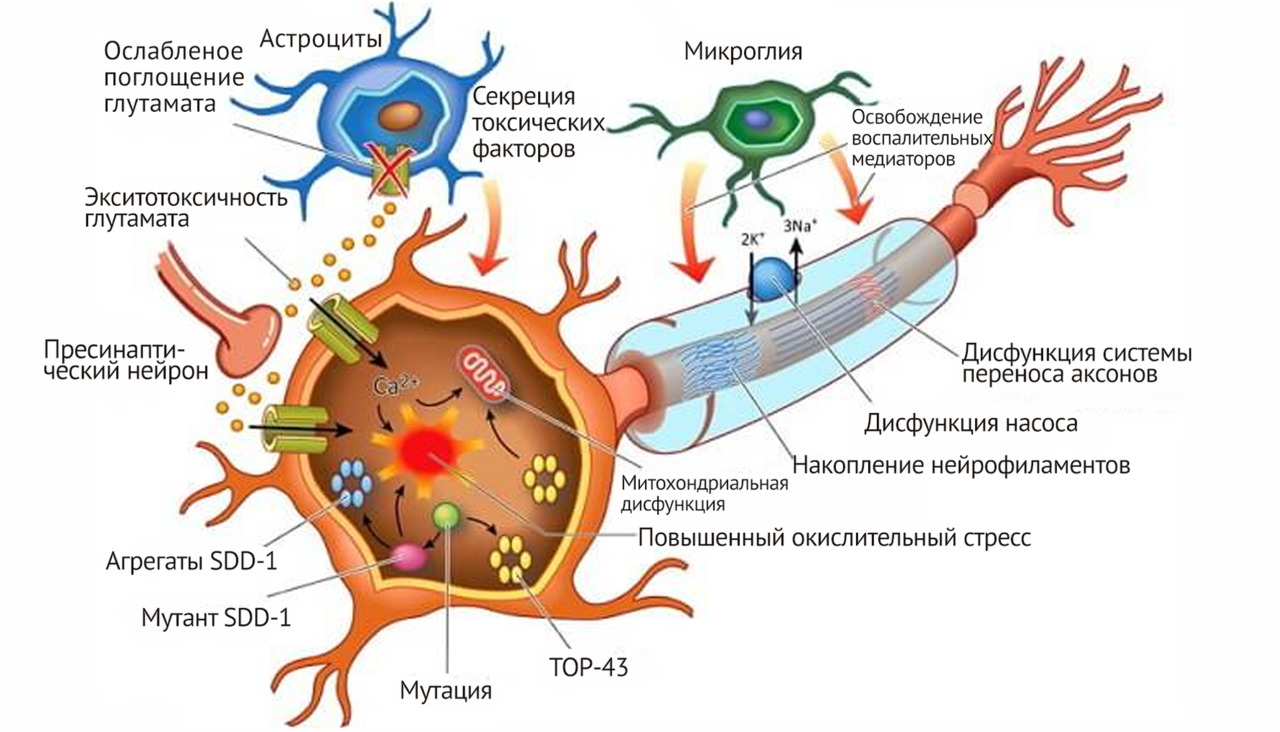
Рис. 2. Схема локальных молекулярных повреждений в двигательном нейроне при боковом амиотрофическом склерозе
Как видно на рисунке 2, локальные молекулярные повреждения в мотонейроне связаны с ослаблением поступления глютамата из астроцитов с формированием экситоксичности глютамата, что приводит к митохондриальной дисфункции в мотонейроне, нарушению обмена внутриклеточного Са2+ и возникновению повышенного окислительного стресса, появлению мутантного фермента СОД-1 и, как следствие, появлению мутаций в других генах ядра нейрона. Эти молекулярно-биологические нарушения приводят к появлению в цитоплазме мотонейрона агрегатов СОД-1, которые усиливают процессы мутагенеза. Освобождение воспалительных медиаторов из клеток микроглии приводит к локальному повреждению структуры аксонов и нарушает их информационную проводимость путем нарушения их цитоскелета.
Все большее число исследований указывают на то, что воспаление в центральной нервной системе, а также повышенная концентрация иммунных клеток, вызывающих воспалительные реакции, обнаруженная в центральной нервной системе пациентов с БАС, являются ключевыми этиопатогенетическими факторами при БАС. Клинические исследования выявили периферическое воспаление в БАС; были обнаружены такие маркеры воспаления, как Т-клетки, цитокины и хемокины. Китайские ученые из Университета Минзу в Китае (Minzu University of China; Пекин) провели систематический обзор и метаанализ 25 исследований, включая исследование 812 пациентов с БАС и 639 человек в контрольной группе. Это было сделано для устранения противоречивых результатов. Сравнивались уровни воспалительных цитокинов пациентов с БАС и пациентов контрольной группы, затем данные клинических исследований объединялись и анализировались. Метаанализ выявил значительную гетерогенность для 8 из 14 цитокинов. Интерлейкин (IL) -2, IL-4, IL-17, фактор роста эндотелия сосудов (VEGF) показали умеренные уровни гетерогенности, тогда как фактор некроза опухоли-α (TNF-α), моноцитарный хемоаттрактантный белок-1 (MCP-1), интерферон гамма (IFNγ), IL-5 показали высокие уровни гетерогенности. Анализ в подгруппах показал, что уровни TNF-α в крови были значительно увеличены у пациентов с БАС по сравнению с пациентами в контрольной группе. Ученые отметили: предыдущие данные показали, что уровни TNF-α, IL-6 и IL-1β в крови повышены у пациентов с болезнью Альцгеймера и болезнью Паркинсона, в то время как рецептор фактора некроза опухоли 1 (TNFR1) увеличен при болезни Паркинсона, что дает представление об общем механизме у различных нейродегенеративных нарушений. Авторы пришли к выводу, что метаанализ впервые использовался для исследования изменений уровней воспалительных цитокинов у пациентов с БАС и показал увеличение уровней TNF-α, TNFR1, IL-1β, IL-6, IL-8 и VEGF в периферической крови у пациентов с БАС по сравнению с пациентами в контрольной группе. В исследовании подчеркивается, что периферические уровни цитокинов могут быть биомаркерами для БАС, что может быть интересным для медицинских работников, стремящихся преодолеть разрыв между постановкой диагноза и появлением симптомов. Исследование было опубликовано 22 августа 2017 г. в журнале Scientific Reports (https://studfiles.net/preview/1818013/page:92/).
Роли дисбаланса трофических факторов в этиопатогенезе БАС в последнее время придается большое значение, и эти инновационные молекулярные исследования оформились в новую теорию трофической недостаточности. Это связано с тем, что нейрональное развитие и выживание нейронов в нервной системе человека зависят от сбалансированной и строго управляемой поддержки, исходящей от трофических факторов. Подобные факторы способны регулировать и управлять такими важными физиологическими процессами, как нейрональная дифференциация, поддержка синапсов, нейрональное выживание, осуществляемое через ингибирование апоптоза, нейрогенез и аксональное прорастание (Korsching, 1993; Boonman, Isacson, 1999; Hou et al., 2008). Кроме того, они создают условия для формирования определенных экологических ниш, пригодных для выживания нейронов (Mudò et al., 2009). Трофическая поддержка имеет первостепенное значение для нейронов спинного мозга и обеспечивается из многочисленных различных клеточных источников, включая астроциты, микроглии, нейроны и эндотелиальные клетки (Ikeda et al., 2001; Béchade et al., 2002; Dugas et al., 2008; Su et al., 2009; Hawryluk et al., 2012). Следовательно, трофическая поддержка рассматривается как перспективная терапевтическая стратегия при нейродегенеративных расстройствах (Kotzbauer, Holtzman, 2006) и способна играть важную роль в клеточных методах лечения, нацеленных на реиннервацию утраченных нейромышечных синапсов (Casella et al., 2010).
БАС (ALS) вызывается выборочной и прогрессирующей потерей спинальных, бульбарных и кортикальных двигательных нейронов, что приводит к необратимому параличу, речевым, глотательным и респираторным нарушениям и, в конечном итоге, гибели индивидуумов при быстром течении болезни. БАС преимущественно спорадичен, и 90% случаев происходят при отсутствии семейной истории заболевания. Тем не менее за последние годы стало очевидно, что многие спорадические случаи сопровождаются изменениями белков, мутации которых были выявлены в семейных случаях, что может по меньшей мере повышать вероятность развития БАС (Deng et al., 2010). Многие из подобных мутаций включают изменения в TAR ДНК-связывающем белке 43 (TDP43) и Fused (FUS) белках в генах саркомы, которые соединяют RNA молекулы (Gordon, 2013; Sreedharan, Brown, 2013), в то время как большинство семейных случаев с паттерном преобладающей аутосомной наследственности вызываются мутациями, происходящими в супероксиддисмутазе 1 (SOD1; Rosen et al., 1993). Трансгенные мыши, выделяющие мутантную форму человеческого SOD1, представляют собой наиболее широко используемую модель для изучения БАС in vivo (Gurney et al., 1994). Трофические факторы рассматриваются в качестве терапевтических мишеней при БАС, когда лечение направлено на восстановление утраченных нейромышечных синапсов и защиту двигательных нейронов от токсичности.
Существует целый ряд хорошо охарактеризованных трофических факторов для ЦНС, таких как нейротрофический фактор головного мозга (BDNF), инсулиноподобный фактор роста 1 (IGF1), цилиарный нейротрофический фактор (CNTF), глиальный нейротрофический фактор (GDNF), фактор нервов (NGF), гормон роста и васкулярный эндотелиальный фактор роста (VEGF). Многие из них тестировались на наличие нейропротекторных свойств в различных экспериментальных моделях БАС. В действительности вирусные векторы, кодирующие факторы роста, являются одним из наиболее эффективных способов сдерживания прогрессирования дегенеративных процессов и пролонгации выживания мышей с БАС (Wang et al., 2002; Kaspar et al., 2003; Azzouz et al., 2004; Dodge et al., 2008).
Недостаток специфических трофических факторов приводит к нарушению дифференцировки двигательных нейронов, а отсутствие какого-либо трофического сигнала приводит к нарушению развития разнообразных субпопуляций двигательных нейронов. Отсутствие глиального нейротрофического фактора видоизменяет местоположение развивающихся двигательных нейронов, иннервирующих конечности (Haase et al., 2002; Kramer et al., 2006), а также выборочно подавляет иннервацию интрафузальных мышечных веретен (Gould et al., 2008). Любопытен факт, что чрезмерная экспрессия данного фактора в мышцах при их развитии вызывает гипериннервацию нейромышечных соединений (Nguyen et al., 1998). В противоположность этому нейротрофический фактор головного мозга может и не оказывать большого влияния на двигательные нейроны, так как, несмотря на то что недостаток этого фактора оказывает существенное негативное воздействие на нормальное развитие сенсорных нейронов, двигательные нейроны способны развиваться без крупных изменений (Ernfors et al., 1994a; Jones et al., 1994). Более того, отдельные субпопуляции двигательных нейронов обнаруживают признаки различной степени чувствительности к недостатку нейротрофинов. Так, отсутствие нейротрофина-3 приводит к полной потере спинальных двигательных нейронов (Ernfors et al., 1994b; Gould et al., 2008), тогда как двигательные нейроны лица остаются незатронутыми, а отсутствие цилиарного нейротрофического фактора CNTF не вызывает изменений в развитии двигательных нейронов на спинальном или краниальном уровнях (DeChiara et al., 1995), хотя потеря рецептора α этого фактора (CNTFRα) приводит к значительному дефициту моторных нейронов, и мыши, испытывающие недостаток в этом рецепторе, погибают в перинатальном периоде (DeChiara et al., 1995). Возможным альтернативным или дополнительным лигандом для этого рецептора может служить димер, образованный кардиотрофиноподобным цитокин/цитокиноподобным фактором 1, истощение которого способно вызывать существенное снижение количества двигательных нейронов (Forger et al., 2003). Также было обнаружено, что отсутствие иных факторов, таких как кардиотрофин-1, вызывает существенную потерю двигательных нейронов (Oppenheim et al., 2001; Forger et al., 2003), а отсутствие инсулиноподобного фактора роста-1 (IGF1) вызывает значительное сокращение численности тригеминальных и лицевых двигательных нейронов (Vicario-Abejón et al., 2004). Наконец, в то время как недостаток фактора эндотелиального роста сосудов (VEGF) носит летальный характер, истощение элемента гипоксической ответной реакции в промотерном отделе гена VEGF вызывает снижение экспрессии этого фактора, что приводит к приобретенной прогрессирующей потере двигательных нейронов в мышиных моделях (Oosthuyse et al., 2001). После этого открытия стало известно, что определенные VEGF гаплотипы (-2578С/A, -1154G/A и -634G/C) передавали повышенную восприимчивость к БАС в работах с людьми, но позднее, при проведении метаанализа с участием более 7000 испытуемых, собранных из как минимум 8 различных популяций, не было выявлено какой-либо взаимосвязи между этими гаплотипами и возникновением БАС (Lambrechts et al., 2009). Кроме того, у больных с БАС не были выявлены мутации ни в элементе гипоксической ответной реакции промотерного отдела VEGF (Gros-Louis et al., 2003), ни в VEGF рецепторе 2 (Brockington et al., 2007).
Нейротрофические факторы оказывают большое воздействие не только на ход развития, они также управляют сохранением и поддержанием, выживанием двигательных нейронов на протяжении длительного времени даже после дифференцировки. Таким образом, они могут запускать активирование эндогенных регенеративных процессов. Помимо синтеза трофических факторов в локальной спинальной микросреде, синаптические мишени двигательных нейронов также играют важную роль в механизмах трофической обратной связи. На самом деле этот факт представляется первостепенным для развития ЦНС, так как при этом трофические элементы тканей мишеней достигают формирующиеся нейроны, что позволяет им преодолевать эндогенно закодированную гибель клеток (Oppenheim, 1991). В случае с двигательными нейронами подобные эффекты преимущественно достигаются при помощи факторов, происходящих из скелетных мышц (Oppenheim et al., 1988; Grieshammer et al., 1998; Kablar and Rudnicki, 1999).
Среди всех трофических факторов, протестированных в экспериментальных моделях БАС, фактор VEGF оказался одним из наиболее действенных защитников двигательных нейронов. Фактор VEGF замечательным образом замедляет прогрессирование заболевания и отмирание двигательных нейронов в семейных (Azzouz et al., 2004; Zheng et al., 2004; Storkebaum et al., 2005; Wang et al., 2007), а также спорадических (Tovar-Y-Romo et al., 2007; Tovar-Y-Romo, Tapia, 2010, 2012) опытных моделях моторной нейродегенерации. Активация VEGF рецептора 2 запускает фосфорилирование внутриклеточных путей, управляемых фосфатидилинозитол-3-киназой (PI3-K), фосфолипазой C-γ и митоген-активированной протеинкиназой (MEK), которые способствуют ингибированию проапоптозных факторов, в том числе Bad (Yu et al., 2005), каспазы 9 (Cardone et al., 1998) и каспазы 3 (Góra-Kupilas, Joško, 2005; Kilic et al., 2006). Активация подобных внутриклеточных сигнальных путей была тщательно изучена применительно к ЦНС (Zachary, 2005). VEGF-зависимая активация PI3-K/Akt считается достаточной для предотвращения моторной нейрональной гибели в семейных моделях БАС in vitro (Li et al., 2003; Koh et al., 2005; Tolosa et al., 2008), а также в опытных моделях эксайтотоксической нейрональной гибели in vitro (Matsuzaki et al., 2001). Более того, активация PI3-K/Akt требуется для выживания двигательных нейронов и аксональной регенерации после спинномозгового поражения (Namikawa et al., 2000). Было доказано, что сигналинг, опосредованный PI3-K, критическим образом связан с защитным воздействием фактора VEGF против AMPA-индуцированной эксайтотоксической спинальной нейродегенерации в условиях in vivo (Tovar-Y-Romo, Tapia, 2010).
VEGF также выступает посредником в нейропротекции, осуществляемой через ингибирование стресс-активируемых протеинкиназ, в том числе p38 митоген-активируемой протеинкиназы. Повышенные уровни фосфорилированной p38 отмечались в моторных нейронах и в глии в моделях семейных случаев БАС на мышах (Tortarolo et al., 2003; Holasek et al., 2005; Veglianese et al., 2006; Dewil et al., 2007), даже в пресимптоматической стадии (Tortarolo et al., 2003). А также p38 является важным фактором для пути клеточной гибели, специфическим для двигательных нейронов (Raoul et al., 2006). Заслуживает внимания то, что ингибирование p38 может предотвращать гибель двигательных нейронов в in vitro семейной модели БАС (Dewil et al., 2007). Несколько групп исследователей доказали, что VEGF может подавлять активацию p38 как в семейных, так и в эксайтотоксичных моделях (Tovar-Y-Romo, Tapia, 2010) спинномозговой нейродегенерации.
Повышенная экспрессия VEGF-индуцирующего гипоксия-индуцированного фактора 1 (HIF-1α), наблюдаемая в спинном мозге, может быть вызвана взаимосвязанными гипоксическими состояниями спинальной микросреды, хотя моторные нейроны не кажутся способными к полномасштабному ответу на повышенный уровень нижележащих эффекторов, таких как VEGF (Sato et al., 2012). Одним из возможных объяснений этого обстоятельства, а также снижения экспрессии VEGF, наблюдаемой у пациентов (Devos et al., 2004), может быть тот факт, что индуцирующие факторы, такие как HIF-1α, не допускаются и исключены из процесса перемещения в ядро, несмотря на то что их концентрации увеличиваются в цитоплазме (Nagara et al., 2013). Подобная неспособность задействовать полную ответную реакцию синтеза VEGF во время гипоксии не носит специфический характер для клеточных типов, и такая неспособность отмечалась в моноцитах, полученных у больных БАС (Moreau et al., 2011).
В отличие от хорошего защитного потенциала фактора VEGF, другие факторы, в том числе BDNF, не оказывали защитное воздействие в разных экспериментальных условиях. Фактор BDNF синтезируется посредством активированной микроглии на ранних стадиях заболевания, когда глиальная ответная реакция преимущественно оказывает противовоспалительное и защитное воздействие, однако выработка этого фактора утрачивается при приобретении микроглией токсических элементов на более поздних стадиях (Liao et al., 2012). Кроме того, BDNF не защищает двигательные нейроны от эксайтотоксичности в экспериментальных моделях in vitro (Fryer et al., 2000) и in vivo (Tovar-Y-Romo, Tapia, 2012). Возможно, это объясняется секвестрацией лиганда, которая происходит за счет усеченной изоформы рецептора высокой аффинности, о котором известно, что он выделяется двигательными нейронами, поскольку устранение этого усеченного рецептора значительно замедляет начало болезни в семейной модели на мышах (Yanpallewar et al., 2012). Несмотря на это, BDNF может оказаться фактором риска для нейронов, либо ввиду того, что он увеличивает их чувствительность к эксайтотоксичности (Fryer et al., 2000), либо за счет механизма активации NADPH оксидазы — фермента (Kim et al., 2002), участвующего в патологии двигательных нейронов посредством разрушения путей выживания, активируемых трофическими факторами (Wu et al., 2006). Было показано, что другие факторы роста также оказывали благотворное воздействие, хотя и в меньшей степени.
Экспрессия фактора GDNF, осуществляемая астроцитами, повышается на фоне спинномозговой ишемии, и это может служить механизмом защиты по отношению к моторным нейронам против эксайтотоксической гибели (Tokumine et al., 2003). Фактор GDNF оказывает свое нейропротекторное воздействие преимущественно на нейрональные сомы, нежели на нервные окончания в нейромышечных синапсах, будучи задействованным непосредственно в спинном мозге (Suzuki et al., 2007). Напротив, при воздействии непосредственно на мышцы фактор GDNF сохраняет мышечно-нервный синапс и стимулирует функцию моторных нейронов и их выживание в семейной модели БАС (Suzuki et al., 2007), что дает основание предположить, что защитные свойства, осуществляемые фактором GDNF, носят довольно ограниченный характер и распространяются в пределах трофического источника. Тем не менее GDNF может ретроградно транспортироваться вдоль моторных нейрональных аксонов (Leitner et al., 1999), что создает условия для изучения маршрута доставки, который мог бы влиять как на сомы, так и на нервные окончания. Любопытно, что у людей с БАС выявляются признаки повышенной экспрессии фактора GDNF в мышцах (Grundström et al., 1999), а сверхэкспрессия данного фактора в мышцах, но не в астроцитах приводит к увеличению продолжительности жизни у мышей с моделью БАС (Mohajeri et al., 1999). Комбинированная терапия с использованием факторов роста может оказаться альтернативой, заслуживающей должного изучения, о чем свидетельствует недавнее сообщение о трансгенной модели БАС на крысах, на которой было показано, что факторы VEGF и GDNF, вводимые через имплантат мезенхимальных стволовых клеток человека, оказывают синергическую защиту нервных синапсов мышц (Krakora et al., 2013).
Что касается фактора CNTF, хотя и сообщалось, что блокировка его экспрессии приводит к потере моторных нейронов (Masu et al., 1993) и развитию двигательных симптомов, подобное воздействие носит сравнительно легкий характер в сравнении с действием, индуцируемым потерей других факторов, в частности VEGF. Интересно также то, что у больных БАС выявляется избирательное снижение экспрессии CNTF в тех отделах ЦНС, которые подверглись негативному воздействию болезни (Anand et al., 1995). Напротив, сывороточные уровни CNTF обычно повышены у больных БАС, особенно у пациентов с формой люмбального начала заболевания (Laaksovirta et al., 2008).
Глава 3. Генетические аспекты бокового амиотрофического склероза
В настоящее время доминирует генетическая теория происхождения БАС. Приблизительно 5–10% случаев заболевания являются семейным БАС (FALS), остальные 90–95% случаев — спорадические БАС (SALS). Известно, что около 20% семейного и 5–7% спорадического БАС связаны с мутациями в гене медь-цинк зависимой супероксиддисмутазы (СОД-1) — фермента, утилизирующего свободные радикалы.
Рис. 3. Единственный каузативный ген, кодирующий супероксиддисмутазу-1 (СОД-1), мутации которого приводят к БАС
К настоящему времени открыто 108 мутаций генов при боковом амиотрофическом склерозе. Все, кроме D90A и D96N, наследуются по аутосомно-доминантному типу. Помимо этого, за последнее десятилетие выявлено несколько генов помимо гена СОД-1, мутации которых могут приводить к развитию БАС: гены белков цитоскелета мотонейрона, гены белков, регулирующих выживание мотонейронов, гены белков митохондриальной дыхательной цепи. Основные описанные генетические локусы при боковом амиотрофическом склерозе представлены в таблице 1 и 2.
На данный момент определены более 22 локусов БАС и 4 локуса БАС + FTD (FTD-ALS — frontotemporal dementia — ALS) (БАС наряду с лобно-височной деменцией), и в большинстве случаев идентифицируются гены, связываемые с возникновением заболевания. Это обозрение подводит итог доступной на сегодня информации, которую удалось собрать по четырем наиболее распространенным генам, связываемым с семейными случаями БАС: SOD1, TARDPB, FUS, и C90RF72. Эти гены привлекли внимание к роли окислительного стресса и РНК-процессинга как патогенных механизмов, способствующих развитию БАС.
⠀
Таблица 1.
Описанные генетические локусы при боковом амиотрофическом склерозе, по данным отечественной литературы
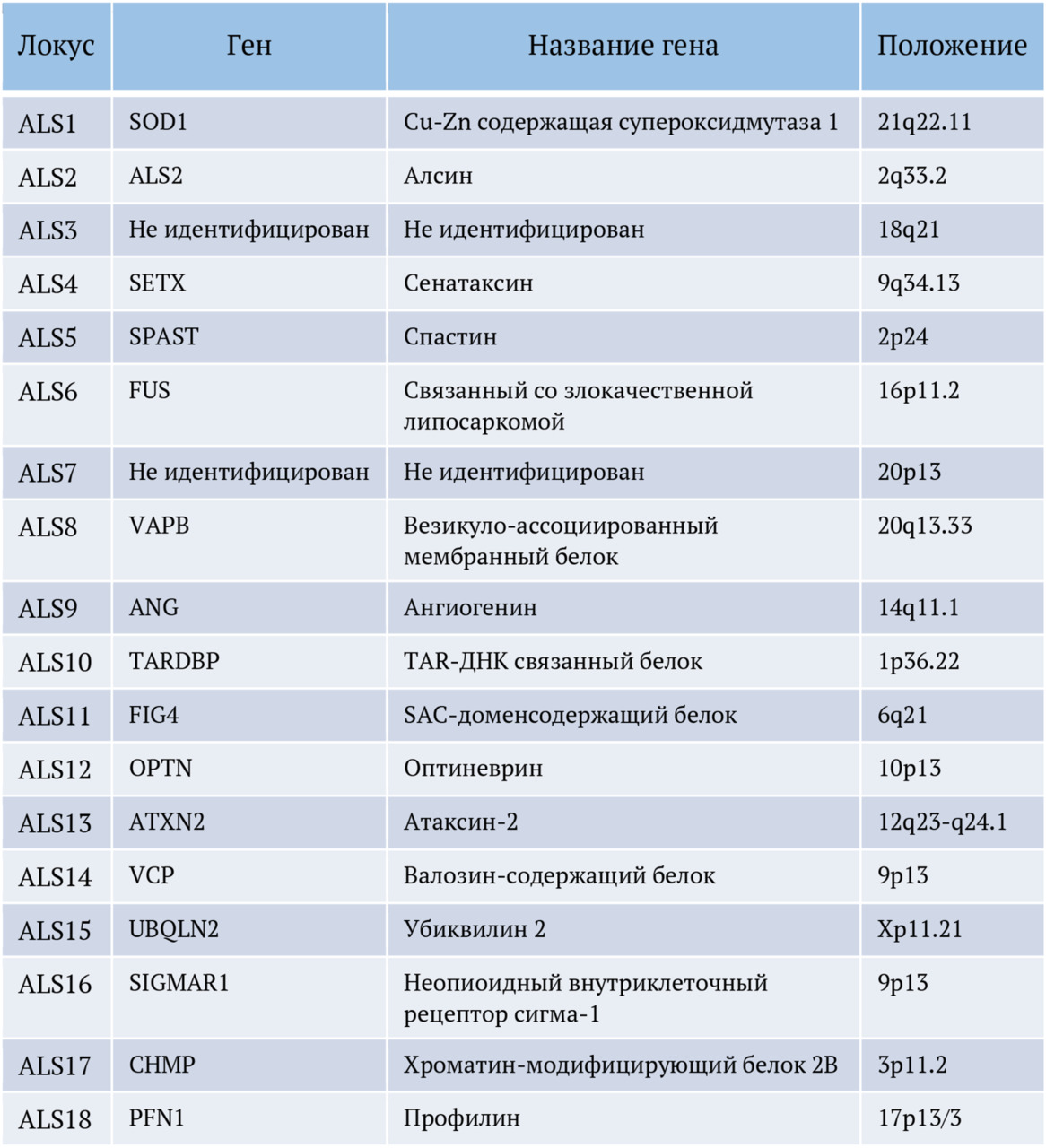
Кроме того, более редкие генетические аллели заключают в себе дополнительные биологические пути, как, например, систему убиквитин-протеасомы (UPS), белковый трафик, а также расстройство цитоскелетной функции.
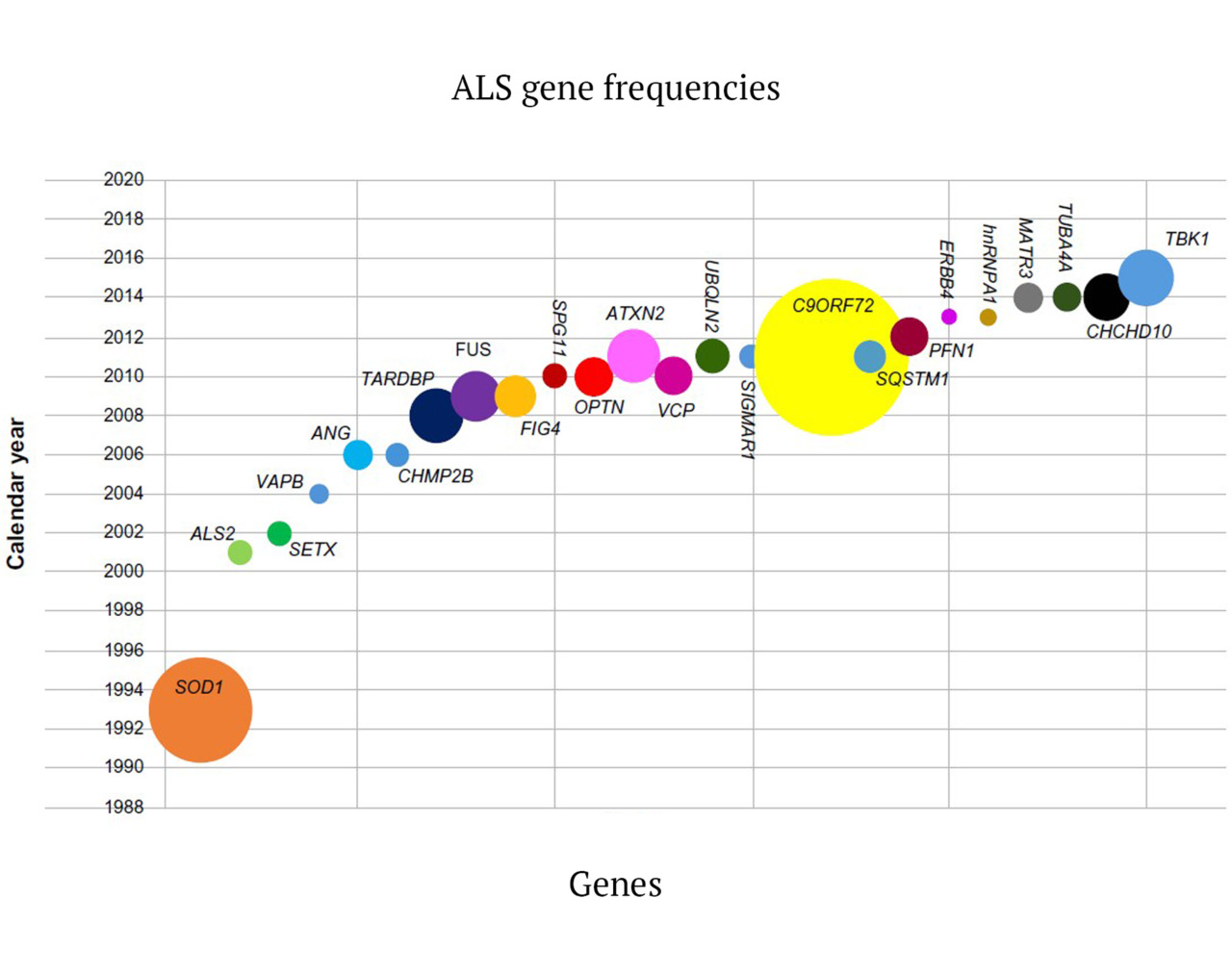
Рис. 4. Исторические вехи и частота открытия различных генов, участвующих в возникновении бокового амиотрофического склероза. Примечание. Каждый ген нанесен на график в соответствии с годом его обнаружения. Размер мутаций в FALS и ALS, как указано в литературе. В тех случаях, когда частоты генов отсутствуют, для иллюстративных целей им присваивается размер круга, эквивалентный 1% (цит. по: Al Sultan et al., 2016)
На представленном рисунке 4 авторы использовали нумерацию локусов, как это приведено в Online Mendelian Inheritance (онлайн каталог фенотипических маркеров у человека) в каталоге фенотипических серий по БАС (PS105400) и FTDALS (PS105550) (Online Mendelian Inheritance in Man, OMIM®, 2016).
БАС1: Cu-Zn дисмутаза супероксида (SOD1). Мутация Cu-Zn супероксиддисмутазы 1 (SOD1) была первой описанной генетической причиной семейных случаев БАС (FALS) (Rosen et al., 1993). Большинство SOD1-мутаций представляют собой аутосомно-доминантную картину, сопровождаемую высокопроникающим паттерном наследственности. Эти мутации преимущественно ассоциируются с возникновением БАС в каких-либо конечностях. Исключением из данного правила является мутация D90A, выявляемая преимущественно у скандинавских популяций, в которых она наследуется в аутосомно-рецессивном виде. Частота SOD1-мутаций варьируется в зависимости от популяций, от 23% в Скандинавии и до 12% в Германии; также мутации определялись в кажущихся спорадических случаях (Andersen, 2006). База данных по БАС (ALSoD database, http://alsod.oip.kcl.ac.uk; Abel et al., 2012) сообщает о 183 мутациях в SOD1, связываемых с болезнью (оценка на ноябрь 2015 г.), большая часть которых представляет собой точечные мутации.
Учитывая, что SOD1 кодирует 153-й аминокислотный белок, это количество мутаций поразительно, причем мутации распределяются по всему гену и оказывают воздействие на целый ряд доменов внутри белка. Данное обстоятельство отличается от некоторых других ассоциированных с БАС мутаций, которые чаще локализуются в пределах определенного мотива выделяемого белка, тем более что неясно, являются ли все заявленные SOD1-мутации действительно патогенными (Felbecker et al., 2010; Marangi, Traynor, 2015). Множественные мутации, происходящие внутри белка, привели также к трудностям определения того, каким образом они отвечают за фенотип болезни. Ген SOD1 является повсеместно экспрессируемым антиоксидантным белком, ускоряющим и катализирующим супероксид свободных радикалов в перекись водорода и кислород. Так как большинство мутантных белков сохраняют эту ферментативную функцию, предполагается, что патогенность оказывает свое воздействие через принцип токсического приобретения функции, хотя точная природа подобной токсичности пока еще остается полностью не изученной. Рассматривался ряд взаимно совместимых патогенных механизмов, в том числе окислительный стресс, экскайтотоксичность, скопление и агрегация белков, нейровоспаление, апоптоз, митохондриальная дисфункция, дисрегуляция аксонального транспорта и стресс эндоплазматического ретикулума (Felbecker et al., 2010; Marangi, Traynor, 2015; Kaur et al., 2016). Мутантные SOD1-белки (mtSOD1) обнаруживают переменные состояния металляции (замещения металлом водорода) и формирования дисульфидной связи, что приводит к тому, что деметаллизированная и развернутая апоформа может проникать в межмембранное пространство митохондрии (пластосомы), вызывая тем самым митохондриальную дисфункцию (cSheng et al1., 2012). Помимо этого, процесс деметаллизации приводит к повышению неустойчивости, и mtSOD1 демонстрируют более высокую предрасположенность к накоплению, чем SOD1 дикого типа.
Таблица 2. Обзор ключевой информации, доступной для локусов ALS и FTDALS, о генах, вовлеченных в развитие бокового амиотрофического склероза (цит. по: Al Sultan et al., 2016)
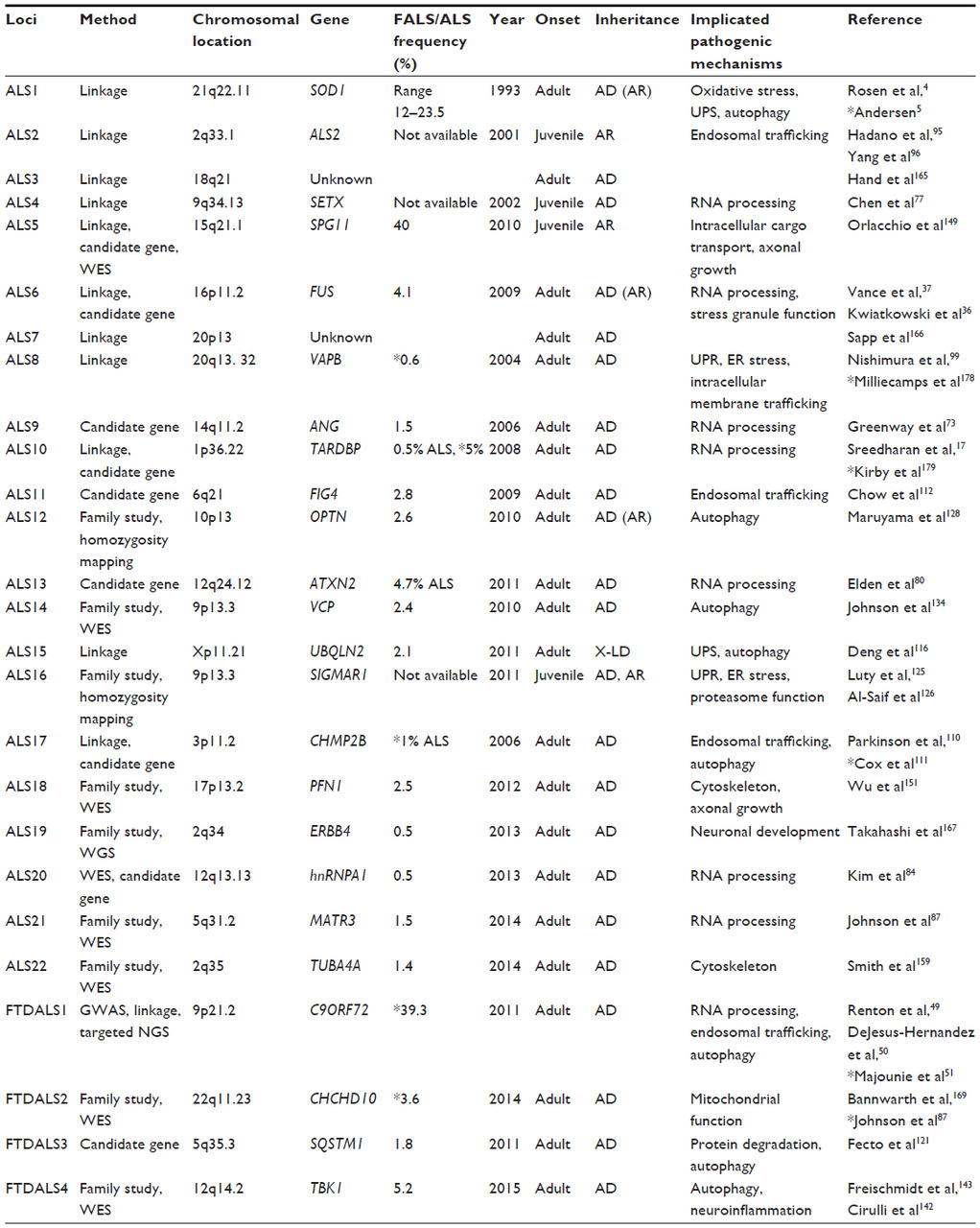
Примечание. Нумерация локусов ALS и FTDALS определяется онлайн-каталогом фенотипических маркеров у человека OMIM®. Частота мутаций в FALS основана на данных из исходных статей или ссылок, помеченных звездочкой (*). В некоторых случаях предоставляется частота в ALS, а не FALS (цит. по: Al Sultan et al., 2016).
Совсем недавно было показано, что мутантный SOD1 (mtSOD1), вместе с неправильно свернутым SOD1 дикого типа, продвигаются от клетки к клетке и инициируют прионо-подобное скопление SOD1 (Grad et al., 2014; Munch, Bertolotti, 2011). Тогда как первоначальное исследование продемонстрировало разрастание неправильно свернутого белка в моделях клеточных культур, спинальные гомогенаты, изъятые из парализованного мышиного мутанта G93A SOD1 и пересаженные в 6-месячную G85R-SOD1:YFP мышиную модель (в которой обычно болезнь не развивалась раньше, чем до 20 мес.), вызывали прогрессирующую болезнь двигательного нейрона в течение 3 месяцев (Ayers et al., 2016).
В обычном виде неправильно свернутые белки изымаются из клеток через механизм действия системы убиквитин-протеасомы (UPS). Однако в БАСе, обусловленном геном SOD1, а также в спорадическом БАСе было показано, что действие системы убиквитин-протеосомы (UPS) нарушается (Kabashi et al., 2012, Kabashi, Durham, 2006). Кроме того, было показано, что MGRN1 (mahogunin ring finger 1), являющийся E3 убиквитин-лигазой, которая катализирует и ускоряет моноубиквитинирование белков и помечает их для деградации при помощи UPS-независимого механизма, сокращается в G93A мышиной модели. Любопытно, однако, что сверхэкспрессия этого белка приводила к сокращению SOD1-токсичности за счет подавления агрегации (скопления) SOD1. Таким образом, терапевтические стратегии по лечению БАС включают повышение очищения от неправильно свернутого SOD1, и индуктор белка теплового шока, arimoclomol, представляет собой один из подобных препаратов, разрабатываемых и изучаемых в настоящее время (Kalmar et al., 2014).
Хотя вначале окислительный стресс считался одним из основных механизмов мутантных SOD1, продолжающиеся исследования патогенного действия SOD1 привлекли и такие факторы, как система убиквитин-протеасомы (UPS), накопление и деградация белков, а также другие аспекты белкового трафика. Подобные пути также связаны с открытием дополнительных генов семейных БАС (FALS) (Раздел «Белковый трафик и гены, имеющие отношение к деградации»).
ALS10/БАС10: TAR ДНК-соединительный белок (TARDBP)
Транзактивный ДНК-связывающий белок 43 (TDP-43) кодируется геном TARDBP на chr1p36.22 (Sreedharan et al., 2008). Ген TARDBP ответственен за 4–5% семейных БАС и примерно 1% спорадических случаев БАС (Millecamps et al., 2010). Мутации в TARDBP наследуются в виде аутосомно-доминантных (AD) проявлений и ассоциируются с классическим клиническим фенотипом БАС. Ген TARDBP кодирует несколько изоформ, из которых преобладает TDP-43. Изоформа TDP-43 является гетерогенным ядерным рибонуклеопротеином (hnRNP), наделенным сигналом ядерной локализации (NLS) и сигналом ядерного экспорта, что способствует челночному перемещению белка между ядром и цитоплазмой. В белке TDP-43 содержатся три последующих домена, два мотива распознавания РНК (RRM1 and RRM2), которые участвуют в соединении РНК и ДНК, а также глицин-обогащенный домен, который необходим для взаимодействия с другими белками и представляет собой локализацию, в которой происходит большинство мутаций (Baralle et al., 2013; Lagier-Tourenne et al., 2010).
Первоначально TDP-43 был идентифицирован как транскрипционный репрессор, соединяющийся с TAR ДНК в вирусе-1 иммунодефицита человека (Ling et al., 2015). После этого было продемонстрировано, что TDP-43 играет роль в РНК-метаболизме, в том числе в РНК-транскрипции, альтернативном сплайсинге, предварительном микроРНК-процессинге, РНК-транспорте и устойчивости матричной РНК (messenger RNA, mRNA) (Scotter et al., 2015). Белок TDP-43 обладает способностью самостоятельно регулировать экспрессию собственных генов за счет соединения с 3´нетранслируемой областью (3´UTR) своего матричного РНК, что порождает неустойчивость и разложение (Ayala et al., 2015). Также TDP-43 соединяется с UG-обогащенными последовательностями в многочисленных последовательностях mRNA (матричной РНК) в целях регулирования сплайсинга (Polymenidou et al., 2011; Sephton et al., 2011; Xiao et al., 2011). Кроме того, недавно была обнаружена новая функция, при которой TDP-43 может подавлять сплайсинг несохраненных (неконсервированных) экзонов, известных как криптические экзоны (Ling et al., 2015). Устранение TDP-43 позволяло этим криптическим экзонам встраиваться в последовательности mRNA (матричной РНК), что затем прерывало транслирование и вызывало нонсенс-опосредованное разрушение. Наконец, TDP-43 также известен в качестве составляющего компонента стрессовых гранул (SGs), хотя и неясно, способствует ли это обстоятельство процессу нейродегенерации (Aulas, Vande Velde, 2015). Роль белка TDP-43 весьма заметна в характерных убиквитиновых цитоплазматических включениях, которые обнаруживаются у больных с БАС и лобно-височной деменцией (Neumann et al., 2006). Примерно 97% больных с семейной и спорадической формами БАС являются положительными для TDP-43 включений в двигательном кортексе и спинном мозге, тем самым подчеркивается значение TDP-43 как основной белковой сигнатуры заболевания, а не только тех белков, переносящих TARDBP-мутации (Sreedharan et al., 2008; Qin et al., 2014).
Потеря ядерной локализации TDP-43 при БАСе хорошо задокументирована, и существуют данные о наступающем в результате этого дефиците сплайсинга в клеточных и животных моделях БАС, а также в образцах, взятых у пациентов (Ling et al., 2015; Highley et al., 2014; De Conti et al., 2015). Кроме утраты ядерной функции, цитоплазматическое приобретение функции также может способствовать нейродегенерации. Модель мыши с мутацией в сигнале ядерной локализации TARDBP человека, что ограничивало TDP-43 до уровня цитоплазмы, продемонстрировала увеличенную экспрессию связанных с транскрипцией и хроматиновой сборкой генов и генов обработки гестона 3´UTR (Amlie-Wolf et al., 2015). Важно отметить, что подобные транскрипционные изменения не наблюдались при добавлении антисмыслового олигомера к нокдаун TDP-43 экспрессии, что, таким образом, поддерживало идею о цитоплазматическом токсическом приобретении функции. Наконец, как и в случае прионоподобного распространения заболевания, описанного при SOD-БАСе, были также получены доказательства того, что TDP-43 олигомеры дикого типа могут распространяться горизонтально от клетки к клетке через микровезикулы, в том числе через лизаты головного мозга больных БАС, а также вертикально по аксонам (Feiler et al., 2015). Таким образом, снижение скоплений подобных мутантных протеинов постепенно становится все более широко используемой терапевтической стратегией.
ALS6/ALS6: соединение с саркомой (FUS)
Ген FUS в chr16p11.2 впервые был идентифицирован как гибридный (соединительный) онкоген липосаркомы. Ген FUS принадлежит семейству FET-белков, и было доказано, что он является hnRNP (гетерогенным ядерным РНП) благодаря своему участию в транскрипционном процессе, транспорте, трафике, альтернативном сплайсинге и обработке микроРНК. Как и в случае с TDP-43, он также присутствует в SGs (стресс-гранулах). По своей структуре FUS состоит из 526 аминокислот, которые образуют N-терминальный домен, обогащенный в глутамин-глицин-серин-тирозине (QGSY), трех аргинин-глицин-глицин обогащенных доменов (RGG-rich domains), RRM, и мотива «цинкового пальца», а также из сигнала ядерного экспорта и NLS (сигнала ядерной локализации), обеспечивающих ядерно-цитоплазматическое челночное курсирование белка (Deng et al., 2014).
Мутации, происходящие в FUS-гене, вначале были выявлены в аутосомно-рецессивном семействе из Кабо Верде, хотя последующий скрининг позволил установить, что FUS также является причинным фактором в аутосомно-доминантном БАС (Kwiatkowski et al., 2009; Vance et al., 2009). Мутации FUS составляют до 4% семейных БАС и 1% спорадических БАС, а большинство мутаций сосредоточено либо в пределах экзонов 3–6, кодирующих QGSY-обогащенный и первый RGG-регион, либо в экзонах 12–15, которые шифруют домен «цинкового пальца», два других RGG-домена и NLS (сигнал ядерной локализации) (Deng et al., 2014). Хотя было продемонстрировано, что в С-терминале мутации являются функциональными, тем не менее мутации, происходящие в экзонах 3–6, чаще выявляются при спорадическом БАС, или они не всегда изолируются от заболевания, что предполагает факт неполной пенетрантности (проявления гена) или непатогенных вариаций.
Ранее очищение от РНК полимеразы II из ядра было показано как приводящее к увеличению цитоплазматического FUS, что позволяет предположить, что FUS играет роль в транскрипции (Zinszner et al., 1998). Впоследствии было показано, что FUS выступает как посредник-медиатор во взаимодействии между РНК полимеразой II и фактором сплайсинга U1 snRNP, тем самым соединяя транскрипцию со сплайсингом (Yu, Reed, 2015). Мутации в FUS приводят к неверной локализации как FUS, так и U1 snRNP в цитоплазме (Yu et al., 2015), а другие РНК-связывающие белки, в том числе SMN1, hnRNPA1, и hnRNP2, также совместно локализуются в mtFUS-скоплениях (Takanashi, Yamaguchi, 2014). К последствиям таких mtFUS-взаимодействий можно отнести дисрегуляцию сплайсинга и увеличение соединения FUS с SMN, что приводит к сокращению в Gem-организмах (Gem bodies), тем самым отражая как потерю, так и приобретение функции, которые осуществляются mtFUS (Sun et al., 2015).
Мутации, происходящие в FUS, также могут передавать патогенность через дополнительные взаимодействия. Было показано, что FUS соединяется с mRNAs и способствует их транспортировке по дендритам (Fujii, Takumi, 2005); впоследствии было показано, что FUS связывается с polyA отростком AMPA рецептора GluA1, управляя его устойчивостью, притом что утрата FUS приводила к сокращению GluA1 (Udagawa et al., 2015). Кроме того, было показано, что FUS транслоцируется с митохондрией, взаимодействуя с митохондриальным белком 60 шаперона теплового шока (mitochondrial chaperone heat shock protein 60, HSP60), что приводит к митохондриальному поражению (Deng et al., 2015). Наконец, mtFUS взаимодействует с Pur-alpha в стресс-гранулах (SGs) и увеличивает фосфорилирование фактора инициации элонгации 2-alpha, соответственно, тем самым блокируя синтез белков (Di Salvio et al., 2015). Однако вклад каждого из подобных взаимодействий относительно патогенеза заболевания подлежит более точному определению.
FTDALS I (лобно-височная деменция-БАС): С90КА72 (С90КА72)
Наиболее распространенная причина семейных случаев БАС на данное время заключается в экспансии интронного GGGGCC-повтора, происходящего в C90RF72. Эта область вначале была определена посредством полногеномных ассоциативных исследований случаев БАС спорадического вида, а также в популяции финских больных БАС (Shatunov et al., 2010; Laaksovirta et al., 2010). Несмотря на то что изначальное секвенирование гена не смогло определить наличие каких-либо точечных мутаций, самый современный метод таргетированного секвенирования этой области установил участок интронного повтора, расположенный между некодирующими экзонами 1a и 1b (Renton et al., 2011; DeJesus-Hernandez et al., 2011). В то время как здоровые испытуемые группы контроля чаще всего обладают менее 10 гексануклеотидными повторами, пациенты БАС обычно переносят 400–2000 повторов. Экспансия повторов была идентифицирована у 37,6% семейных БАС и 6,3% спорадических БАС, а также в диапазоне до 25,1% случаев лобно-височной деменции (Majounie et al., 2012). В этой связи не удивляет тот факт, что наиболее существенный клинический фенотип, ассоциирующийся с этим генетическим подтипом, состоит в повышенной частоте случаев семейной истории лобно-височной деменции. Помимо того, есть доказательства, что большее количество случаев бульбарного возникновения ассоциируются с БАС, который связан с геном C90RF72 (до 44%, в сравнении с 25–26% в не связанных с геном C90RF72 случаях БАС), а некоторые исследования также сообщают о более раннем возрасте возникновения заболевания (на 1,8–5,0 лет) и его более короткой продолжительности (на 5,7–12,0 мес.) (Cooper-Knock et al., 2015).
Функция C90RF72-гена в настоящее время изучается, хотя структурный анализ позволил установить, что функция этого гена схожа с функцией GDP/GTP-факторов обмена, регулирующих Rab-GTP-азы и может регулировать везикулярный трафик (Levine et al., 2013). Дальнейшее исследование продемонстрировало, каким образом C90RF72 совместно локализовался с Rab-белками, участвующими в аутофагии и эндосомальном трафике (Farg et al., 2014). Хотя механизм действия функции в настоящее время устанавливается, были предложены несколько гипотез относительно того, каким образом интронный гексануклеотидный повтор может вызывать нейродегенерацию: 1) гаплонедостаточность, 2) РНК-токсичность, 3) белковая токсичность дипептидных повторов.
Гаплонедостаточность
Сниженные уровни C90RF72-транскрипта были замечены у больных с экспансией повторов по сравнению с контрольными испытуемыми, и гипотеза о гаплонедостаточности получила подтверждение, когда нокдаун (выключение) гомолога C9orf72, смоделированного у данио (zebrafish), привел к аксональной дегенерации (Ciura et al., 2013). В противоположность этому, в условной модели у C9orf72 нокаутированной мыши, у которой C9orf72 был специально удален из нейрональных клеток, не были получены какие-либо данные, свидетельствующие о нейродегенеративном фенотипе (Koppers et al., 2015). Тем не менее систематическое изучение уровней экспрессии трех C90RF72-транскриптов (вариант 1 = экзон 1а, 2–5; вариант 2 = экзон 1b, 2–11; вариант 3 = экзон 1а, 2–11) показало существенно сокращенную экспрессию вариантов 1 и 2, отмечавшуюся в мозжечке и лобном отделе коры (фронтальном кортексе) переносчиков экспансии C90RF72, и наблюдалось корреляционное соответствие между более высоким уровнем экспрессии варианта 1 и выживаемостью (van Blitterswijk et al., 2015). Данный факт предполагает, что стратегии антисмысловых олигомеров должны избегать сокращения уровней экспрессии C90RF72.
РНК-токсичность
T.F. Gendron и команда определили, что локусы (очаги) РНК расположены преимущественно в ядре и, периодически, в цитоплазме двигательных нейронов, и отметили, что эти локусы состоят как из смысловых, так и из антисмысловых РНК (Gendron et al., 2013). Как оказалось, наличие антисмысловых РНК-очагов находится в корреляционном соответствии с неверной локализацией TDP-43, но не с белковой токсичностью дипептидных повторов (DPRs) (Gendron et al., 2013; Cooper-Knock et al., 2015). Считается, что последовательность повторов формирует G-квадруплексные (учетверенные) структуры внутри клетки. Многие РНК-связывающие белки совместно локализуются с РНК-очагами, теоретически изолируя их из клетки и прерывая их РНК-процессинговые (обрабатывающие) функции (Cooper-Knock et al., 2014, Lee et al., 2013). Это обстоятельство может лежать в основе существенной дисрегуляции РНК-сплайсинга, которая отмечается при экспансии, когда более высокий разрыв (прерывание) заметен у больных с более короткой выживаемостью (Cooper-Knock et al., 2015, Prudencio et al., 2015). Однако очаги РНК отмечаются и в фибробластах, полученных от бессимптомных пациентов (Cooper-Knock et al., 2014, Lagier-Tourenne et al., 2013), и в моделях БAC на трансгенных мышах, содержащих расширенную аллель; тогда как очаги РНК и дипептидные повторы воспроизводят нейропатологию БАС, сведения, которые могли бы подтвердить наличие нейродегенерации, отсутствуют (Peters et al., 2015, O’Rourke et al., 2015). Это противоречит данным по мышиной модели, экспрессирующей 66-повторную G4C2-экспансию конкретно в ЦНС, что продемонстрировало нейропатологические, поведенческие и двигательные нарушения уже к 6 месяцам (Chew et al., 2015).
Белки дипептидного повтора
Наконец, было показано, что экспансия GGGGCC-повторов подлежала повтор-ассоциированной не-анти-тимоцитарной глобулиновой трансляции (repeat-associated non-ATG translation) (Mori et al., 2013). Как смысловые, так и антисмысловые РНК транслируются, образуя белки дипептидных повторов, состоящие из poly-GA, -GP, -GR, -PA и -PR (с poly-GP, продуцируемой как из антисмысловых, так и из смысловых РНК) (Gendron et al., 2013; Mori et al., 2013). Эти белки дипептидного повтора наблюдаются в виде скопления внутри нейрональных цитоплазматических включений и нейрональных внутриядерных включений в двигательной коре мозга, мозжечке, гиппокампе и спинном мозге и положительно маркируются к убиквитину и p62. В недавнее время антитела, восставшие против каждого из белков дипептидного повтора, продемонстрировали лишь небольшую корреляционную зависимость между распространением/грузом дипептидных повторов (DPR) и клиническим фенотипом (Mackenzie et al., 2015; Davidson et al., 2015), что, по мнению авторов, говорит против того, чтобы рассматривать скопление и агрегацию дипептидных повторов как главный патогенный механизм. Это противоречит работе, использовавшей модель дрозофилы, в которой экспрессия дипептидных повторов порождала нейродегенерацию в глазе насекомого (Mizielinska et al., 2014).
В итоге появляется все большее количество данных, говорящих в пользу некоторой формы РНК-дисрегуляции как фактора, способствующего БАС, обусловленному действием C90RF72. Однако пока различные клеточные и животные модели, применяющие разнообразные генетические конструкции, генерируют противоречивые результаты относительно вклада каждой из трех гипотез в развитие заболевания, более точные механизмы все еще подлежат полноценному разъяснению. Прерывание (разрыв) C90RF72-белковой функции в эндосомальном трафике может также представлять собой способствующий фактор.
Другие РНК-процессирующие гены, причастные к БАС
До определения генов TARDBP и FUS как генов, связанных с возникновением БАС, уже была доказана причастность к заболеванию двух РНК-процессирующих генов: ангиогенина (ANG) и сенатаксина (SETX). Впоследствии мутации в hnRNPA1 и matrin 3 (MATR3) были установлены методом полноэкзомного секвенирования, и атаксин 2 (ATXN2) определили как фактор риска.
ALS9/БАС9: ангиогенин (ANG)
После идентификации ANG однонуклеотидных полиморфизмов rs11701, представленных в большом количестве и превалирующих в случаях БАС по Шотландии и Ирландии, скрининг на ANG установил 7 миссенс-мутаций (с изменением смысла) в 15 случаях БАС, из которых 4 являлись семейной формой заболевания (FALS) (1,54%), а 11 относились к спорадическому БАС (SALS) (0,80%) (Greenway et al., 2006). Ген ангиогенин (ANG) является членом суперсемейства панкреатической рибонуклеазы и обладает нейропротекторными свойствами, хотя в mtANG эти свойства нарушаются (Subramanian et al., 2008). В то время как многочисленные мутации были определены, p.K17I не всегда демонстрировал сегрегацию заболевания. Тем не менее метаанализ показал, что у представителей белой европеоидной расы, носителей этой аллели, риск возникновения БАС выше в 1?65 раза, и он увеличивался десятикратно в семейной форме БАС (Pan et al., 2015). Оказалось, что ангиогенин (ANG) индуцирует сборку стресс-гранул (SGs) (Ivanov et al., 2014). Любопытно, что подобная индукция может ингибироваться G-квадриплексными структурами, которые формируются с помощью G4C2 C90RF72 расширенного повтора, тем самым устанавливая взаимосвязь между C90RF72 и ANG.
ALS4/БАС4: сенатаксин (SETX)
Мутации в SETX ассоциируются с подростковым стартом БАС, сопровождаемым ослаблением дистальных мышц и отсутствием бульбарных или чувствительных симптомов. У больных обычно отмечается длительное и медленное прогрессирование болезни, при котором сохраняется сравнительно нормальная продолжительность жизненного цикла (Chen et al., 2004; Hirano et al., 2011). Редкие мутации аутосомно-доминантного вида в SETX имеют место при БАСе, тогда как рецессивные SETX-мутации ассоциируются с атаксия-глазодвигательной апраксией-2 (Hirano et al., 2011). Механизмы, посредством которых SETX-варианты приводят к БАС, остаются неизвестными; тем не менее SETX шифрует ДНК/РНК-геликазный белок, который играет роль в восстановлении ДНК в ответ на окислительный стресс. Ген SETX также взаимодействует с РНК-процессирующими белками, регулирующими транскрипцию и pre-mРНК процессинг, что позволяет выдвинуть теорию о том, что причина дегенерации двигательного нейрона, обусловленной мутациями в SETX, может являться результатом патологического РНК-процессинга (Skourti-Stathaki et al., 2011).
ALS13/БАС13: атаксин 2 (ATXN2)
Более 36 повторов CAG внутри ATXN2 способны вызвать спинально-церебеллярную атаксию 2; однако было замечено, что промежуточные повторы в диапазоне 27–33 тесным образом ассоциируются с БАС, и таким образом установлено, что ATXN2 модифицирует токсичность TDP-43 в дрожжах (Elden et al., 2010). Белок ATXN2 представляет собой РНК-связующий белок, участвующий в РНК-процессинге и локализующийся в эндоплазматическом ретикулуме, аппарате Гольджи и SGs; ATXN2 также взаимодействует с FUS, и промежуточные экспансии обостряют мутантный фенотип FUS в клеточных моделях (Farg et al., 2013). Недавний метаанализ более 6000 больных БАС и 7000 контрольных испытуемых установил, что длины повторов в диапазоне 25–28 обладали в действительности протекторными свойствами, тогда как существенный риск связан с CAG-повторами в диапазоне 31–33 (Neuenschwander et al., 2014). Данное наблюдение поддерживается итальянским исследованием, в котором дополнительно <31 повтора соотносились со спинальным стартом БАС и сокращенной выживаемостью (Borghero et al., 2015).
ALS20/БАС20: гетерогенный ядерный рибонуклеопротеин A I (hnRNPA I)
После доказательства с использованием методов секвенирования экзомов того, что hnRNPA 1 и hnRNPA2B1 мутации являются причинными в семействах мультисистемной протеинопатии, эти гены были специально изучены в 212 случаях семейного БАС, по которым метод экзомного секвенирования был доступен (Kim et al., 2013). Был выявлен отдельный случай, сопровождавшийся мутацией в hnRNPA1. Любопытно, что hnRNPA1 и A2/B1 являются известными взаимодействующими партнерами TDP-43, а hnRNPA1 также взаимодействует с убиквилином-2 (Gilpin et al., 2015). В двигательных нейронах БАС существует утрата интенсивного hnRNPA1 ядерного окрашивания, что также соотносится с ядерной потерей в TDP-43, хотя и hnRNPA1 не был замечен как совместно локализующийся с TDP-43 во включениях, сформированных в виде клубка (Honda et al., 2015). Однако скрининг 113 итальянцев с семейным БАС и 135 голландцев с семейной формой, а также 1084 голландцев со спорадической формой не выявил какие-либо hnRNPA1 мутации, что приводит нас к выводу о том, что это очень редкая причина возникновения семейной формы БАС.
ALS21/БАС21: матрин 3 (MATR3)
Экзосомное секвенирование крупных родословных генеалогий привело к идентификации мутации в MATR3-гене; предыдущее семейство, переносящее мутацию в MATR3 и первоначально диагностированное как аутосомно-доминантная дистальная, асимметричная миопатия с парезом голосовых связок, подверглось переоценке, и заново вынесенный диагноз заключался в заболевании БАС (Johnson et al., 2014). Дальнейший скрининг случаев БАС среди итальянцев и британцев позволил идентифицировать еще две мутации: одну семейную БАС (FALS) и одну спорадическую (SALS). В целом MATR3 является РНК/ДНК-связующим белком, взаимодействующим с TDP-43; в то время как p.S85C мутация усиливает данное взаимодействие, две другие мутации, p.F115C и p.T22A, не ведут к его усилению. Тем не менее это различие может лежать в основе медленного прогрессирования заболевания в том семействе, которое является переносчиком p.S85C-мутации. Хотя последующие мутации не были обнаружены в 372 случаях семейной БАС из Франции, Тайваня, Австралии и французской Канады, 4 мутации были выявлены в случаях, казавши [ся спорадическим БАС (3 у французских канадцев и 1 у гражданина Тайваня).
Гены семейства FET
ТАТА box связующий протеин-ассоциированный фактор 15 (TAF15) и регион 1 точки разрыва саркомы Юинга (EWSR1) представляют собой РНК-связующие белки, которые, наряду с FUS, образуют FET-семейство белков. Во всех трех содержатся прионоподобные домены, и эта особенность используется для ранжирования потенциальных РНК-связующих белков как причастных к развитию БАС вслед за функциональным дрожжевым скринингом (Couthouis et al., 2011). Скрининг TAF15 идентифицировал пять миссенс-вариантов (с изменением смысла) в 1262 случаях БАС, в то время как скрининг EWSR1 идентифицировал 2 потенциальных мутации у 817 случаев БАС (Couthouis et al., 2012). Хотя эти варианты отсутствовали у испытуемых контрольной группы, тем не менее их удалось определить у больных со спорадическим БАС, таким образом сегрегация (разъединение) не могла быть продемонстрирована. Однако как TAF15, так и EWSR1-белки обнаруживают цитоплазматическую неверную локализацию в спорадических видах БАС. В недавнее время методом полногеномного секвенирования (WGS) была выявлена мутация EWSR1 в группе монозиготных (однояйцевых) близнецов, неконкордантных (не предрасположенных) к развитию БАС, что позволяет предположить наличие дополнительных факторов, воздействующих на заболевание (Meltz Steinberg et al., 2015).
Белковый трафик и гены, имеющие отношение к деградации
Начиная с идентификации первого гена ALS2, характерного для аутосомно-рецессивного вида (AR) БАС, наряду с отличительными убиквитинированными включениями, происходящими в двигательных нейронах при БАС, дисрегуляция белкового трафика и деградация (разрушение) белков оказываются причастны к процессу заболевания. К мутациям, происходящим в генах, участвующих в эндосомальном транспорте, относятся алсин (alsin, ALS2), белок B, связанный с везикуло-ассоциированным мембранным белком (VAMP-associated protein B, VAPB), хроматин-модифицирующий белок 2B (CHMP2B) и 5-фосфатаза фосфоинозитида (FIG4); к генам, участвующим в системе убиквитин-протеасомы (UPS), относятся убиквилин 2 (ubiquilin 2, UBQLN2), секвестосома 1 (SQSTM1) и сигма-нонопиоидный внутриклеточный рецептор 1 (SIGMAIR1), а аутофагия преимущественно обусловлена мутациями в оптинейрине (OPTN), валозин-содержащем белке (VCP), и tank-соединительной киназе 1 (TBK1). Существует некоторое перекрытие между этими тремя биологическими путями, которые также имеют отношение к БАС, обусловленному действием SOD1 и C90RF72.
ALS 2/БАС2: алсин (ALS2)
Ген алсин (ALS2) изначально был выявлен при помощи анализа групп сцепления генных локусов (linkage analysis) в родственных семействах Туниса и Саудовской Аравии (Yang et al., 2001; Hadano et al., 2015). Большинство мутаций приводило к белковому усечению, что позволяет сделать предположение о потере функции. Считается, что алсин играет роль в активации Rab5 GTP-аз. Значение Rab5 первостепенно для эндосомального трафика, а в мышиных моделях с нокаутом алсина нейроны показали увеличенное эндосомальное слияние и деградацию, но сниженный уровень подвижности (Lai et al., 2009; Lai et al., 2006). Одним из компонентов эндосомы является AMPA-рецептор GluR2, значения которого сокращаются у мышиных моделей с нокаутом алсина (Lai et al., 2006).
ALS8/БАС8: белок B (VAPB), ассоциированный с везикуло-ассоциированным мембранным белком (VAMP)
Анализ групп сцепления крупного семейства из Бразилии впервые определил белок VAPB как причинный ген БАС (Nishimura et al., 2004), а p.P56S-мутация была выявлена у многочисленных семей из Бразилии, что указывает на какую-то общую основу (Nishimura et al., 2005). Были сообщения о дополнительных мутациях, хотя и не все вариации были изолированными от заболевания (Nishimura et al., 2005; Chen et al., 2010; Kabashi et al., 2013; van Blitterswijk et al., 2012). Белок VAPB является интегральным белком 2-го типа мембраны эндоплазматического ретикулума, который принимает участие во внутриклеточном трафике и ответной реакции несвернутого белка (Lev et al., 2008), а также в регулировании взаимодействия между эндоплазматическим ретикулумом и митохондрией (Stoica et al., 2014). Однако p.P56S-мутантный белок не вызывает ответную реакцию несвернутого белка, перемены в поглощении кальция в митохондрии и нарушение антероградного аксонального транспорта митохондрии (Kanekura et al., 2006; De Vos et al., 2012; Morotz et al., 2012).
ALS17/БАС17: хроматин-модифицирующий белок 2B (CHMP2B)
Мутации в CHMP2B вначале были определены в 2 вероятных случаях семейного БАС и в последующих 3 случаях спорадического БАС; большинство из мутаций выявляли преобладающий фенотип нижнего двигательного нейрона (Parkinson et al., 2006; Cox et al., 2010). Белок CHMP2B является компонентом ESCRT-III системы эндосомального трафика, сортирующей «грузы» в мультивезикулярные тельца. Не так давно 4 новейших мутации были выявлены в случаях БАС, кажущихся спорадическими, и они располагались в домене, который необходим для формирования мультивезикулярных телец (van Blitterswijk et al., 2012). В клеточных моделях мутантный CHMP2B приводил к формированию крупных вакуолей и возрастанию LC3-II маркера аутофагии, подразумевая дисрегуляцию аутофагии как механизм, способствующий развитию БАС.
ALSI I: фосфоинозитидная 5-фосфатаза (FIG4)
Мутации в FIG4 вначале определялись как являющиеся причинным фактором в болезни Шарко — Мари — Тута по типу 4J, хотя у одной семьи наблюдался клинический фенотип, напоминающий БАС. Скрининг семейных и спорадических случаев БАС определил 9 вариантов, 6 из которых показывали нарушение в функции дрожжей (Chow et al., 2009). FIG4, также известный как SAC3, регулирует уровни комплекса PI (3,5) P2 и тем самым контролирует ретроградный трафик энодосомальных везикул в область Гольджи. Мутантные белки показывали потерю фосфатазной активности, неправильную локализацию и неспособность соединяться с PI (Online Mendelian Inheritance in Man, 2016; Andersen, 2006) P2-комплексом. Дальнейший скрининг популяций больных из Италии и Тайваня не смог обнаружить какие-либо новейшие варианты, хотя лишь 80 спорадических БАС и 15 семейных БАС проходили скрининг в каждом исследовании (Tsai et al., 2011; Verdiani et al., 2013). Оценка патологии по переносчикам мутации не проводилась; однако не было показано, что FIG4 локализуется в спорадическом виде БАС неправильно (Kon et al., 2014).
ALS15/БАС15: убиквилин 2 (UBQLN2)
При анализе групп сцепления генных локусов крупного семейства, насчитывающего несколько поколений, был идентифицирован UBQLN2. Скрининг дополнительных случаев семейного БАС, исключавших передачу от мужчины к мужчине, позволил обнаружить еще 4 мутации; они все были расположены в PXX-области повтора белка (Deng et al., 2011). Дополнительный скрининг определил последующие варианты, примыкающие к PXX-области повторов или расположенные в ней (Williams et al., 2012; Gellera et al., 2013). Было показано, что мутации приводят к прерыванию и разрушению пути деградации белков, которое осуществляется через недостаточное соединение с протеасомой (Chang, Monteiro, 2015) и вызывает неправильную локализацию OPTN из Rab-11-позитивных эндосомальных везикул (Osaka et al., 2015), а также потенциально может повредить РНК-метаболизм через утрату соединения UBQLN2 с hnRNP-белками, в том числе с hnRNPA1 (Gilpin et al., 2015).
FTDALS3: секвестосома 1 (SQSTM I)
Ген SQSTM1, или p62, представляет собой убиквитин-соединительный белок, который играет роль в деградации белка, осуществляемой за счет протеасомы и аутофагии, и его можно найти в характерных убиквитиновых включениях у больных БАС. Скрининг этого гена выявил множественные мутации как в семейных, так и в спорадических случаях БАС (Fecto et al., 2011). Дальнейшие мутации были замечены у больных БАС, некоторые в сочетании с костной болезнью Пеждета, о которой известно, что она также индуцируется мутациями в SQSTM1 (Teyssou et al., 2013; Kwok et al., 2014). В данио-модели, в которой нокаутирован эндогенный SQSTM1, изучаемая особь продемонстрировала поведенческие и аксональные патологии, а также прерванную аутофагию, что было зафиксировано увеличением уровней mTOR (Lattante et al., 2015). Человеческий SQSTM1 был в состоянии спасать фенотип, но часто обнаруживаемая мутация, p.P392L, не могла делать это.
ALS16/БАС16: сигма нонопиоидный внутриклеточный рецептор 1 (SIGMAR 1)
Изначально 3-UTR варианты были выявлены в нескольких семействах с аутосомно-доминантной лобно-височной деменцией, сопровождаемой БАС (FTD-ALS), или c лобно-височными деменциями (FTD), что позволило предположить, что патогенность передавалась через механизм изменения устойчивости мРНК (Luty et al., 2010). Однако, используя метод картирования гомозиготности, впоследствии была выявлена миссенс-мутация в SIGMAR1, которая изолировалась в крупном генетически родственном семействе с признаками ювенильного (юношеского) БАС аутосомно-рецессивного вида (ARJALS) (Al-Saif et al., 2011). SIGMAR1 является шапероном эндоплазматического ретикулума и производной единицей лигандо-регулируемого кальциевого канала и способствует транспорту митоходриального кальция через IP3-рецептор; мутация в SIGMAR1 вызывает формирование цитоплазматических скоплений, сокращение продуцирования ATP и последующее понижение протеасомной активности (Fukunaga et al., 2015). Однако все еще предстоит точно определить, способствует ли SIGMAR1 возникновению БАС аутосомно-доминантного вида.
ALS12/БАС12: оптинейрин (OPNT)
Мутации в OPNT вначале были определены методом картирования гомозиготности генетически родственных японских семейств с аутосомно-рецессивным БАС, что позволило выявить гомозиготную экзонную делецию (стирание) и гомозиготную нонсенс-мутацию (Maruyama et al., 2010). Последующий скрининг случаев семейных БАС определил 2 аутосомно-доминантных семейства, которые были гетерозиготными по отношению к миссенс-мутации. OPNT выполняет свою функцию через белок-белковые взаимодействия: он привязывается к убиквитину и UBQLN2, он является рецептором аутофагии (способствуя рекрутингу грузов к аутофагосомам), он необходим для выстраивания Гольджи (Kamada et al., 2014), и он также регулирует NF-kB сигналирование (Bansal et al., 2015). Последующий скрининг определил дополнительные гетерозиготные мутации в случаях спорадических БАС (van Blitterswijk et al., 2012) и аутосомно-рецессивных БАС (Beeldman et al., 2015; Goldstein et al., 2016).
ALS14/БАС14: валозин-содержащий белок (VCP)
Метод экзомного секвенирования итальянской семьи, насчитывающей 4 поколения, первоначально предположил наличие мутации валозин-содержащего белка (VCP) в качестве причины развития БАС (Johnson et al., 2010). Впоследствии еще 4 варианта были идентифицированы в случаях семейных БАС, предоставляя тем самым дальнейшие подтверждения того, что VCP ассоциируется и связан с БАС. Валозин-содержащий белок (VCP) представляет собой ААА+ (расширенное семейство АТФаз, связанное с различными клеточными действиями) белок АТФаз (ATPase), который вовлечен в целый ряд клеточных функций, включая регулирование протеасомальной деградации убиквитинового белка в мультимерных комплексах и таргетинг субстратов в аутофагосомы (Meyer, Weihl, 2014). Несмотря на то что скрининг не смог выявить какие-либо мутации валозин-содержащего белка в некоторых популяциях (Miller et al., 2012; Williams et al., 2012; Tiloca et al., 2012), потенциальные мутации были выявлены в других популяциях, а также в случаях спорадического БАС (Koppers et al., 2012; Abramzon et al., 2012). Кроме того, мутации VCP (валозин-содержащего белка) ассоциируются с миопатией телец-включений, сопровождаемой ранним возникновением болезни Педжета и лобно-височной деменцией, а фибробласты, изолированные у больных, показали митохондриальное разъединение и сокращение выработки АТФ (Bartolome et al., 2013); эта особенность также отмечается при SIGMAR1-мутациях.
БАС с лобно-височной деменцией/FTDALS4: TANK-связывающая киназа (TBK 1)
Мутации в TBK1 вначале были определены посредством метода секвенирования экзомов в 2874 случаях БАС; преобладающие варианты были обнаружены в 1,097% случаев, а мутации с потерей функции — в 0,382% (Cirulli et al., 2015). За этим вскоре последовало второе исследование, в котором секвенирование 252 случаев семейного БАС установило 9 потерь функции и 4 миссенс-мутации (Cirulli et al., 2015). Мутации затем были обнаружены в случаях БАС, БАС-лобно-височной деменции (FTDALS) и лобно-височной деменции (FTD) (Gijselinck et al., 2015; Le Ber et al., 2015). Танк-связующая киназа (TBK1) играет роль как во врожденном иммунитете, так и в NF-kB сигналировании, а также в аутофагии. TANK-связующая киназа TBK1 соединяется и фосфорилирует БАС-ассоциированные белки OPTN и SQSTM 1, при этом показано, что мутанты TBK1 больше не соединяются с белком OPTN (Freischmidt et al., 2015).
Нарушение аксональной транспортировки
и цитоскелетная дисфункция
Нейроны являются чрезвычайно крупными клетками, которым необходим транспорт органелл, протеинов и РНК, получаемых из тел клеток и доставляемых по аксонам. Молекулярные моторы, такие как кинезины и динеин, направляют подобные «грузы» в микротрубки, чтобы осуществлять, соответственно, антероградную и ретроградную транспортировку. Хотя мутация, происходящая в p150 динактиновой подгруппе (p150 dynactin subunit), в мышиной модели привела к выработке нейродегенеративного фенотипа, мутации в этом гене не были обнаружены в БАС человека (Ahmad-Annuar et al., 2003; Vilarino-Guell et al., 2009). Однако метод экзосомного секвенирования позволил выявить некоторое количество цитоскелетных генов, в которых мутации были заявлены как являющиеся причиной БАС.
ALS5/БАС5: спатаксин (SPG II)
Полноэкзомное секвенирование (WES) двух пораженных заболеванием сиблингов (родных братьев или сестер), происходящих из неединокровного семейства, с подтвержденным ювенильным БАС аутосомно-рецессивного типа, установил лишь один ген, SPG11, в котором варианты были обнаружены в компаундном гетерозиготном состоянии (Daoud et al., 2012). Причастность гена наследуемого спастического парапареза, обычно ассоциируемого с белком теплового шока (HSP) с признаками истончения мозолистого тела черепа, ранее связывалась со скринингом гена-кандидата SPG11, в котором были выявлены мутации у 10 семейств с ARJALS (Orlacchio et al., 2010). Несмотря на то что точная функция белка не установлена, происходящие из индуцированных плюрипотентных стволовых клеток (iPSC-derived) нейрональные клетки с SPG11-мутациями демонстрировали наличие белка, совместно локализованного с цитоскелетом, а мутации вызывали аксональную неустойчивость и нарушение аксонального транспорта (Perez-Branguli et al., 2014).
ALS18/БА18: профилин I (PFN I)
Два БАС-семейства, насчитывающие несколько поколений, были установлены как переносящие мутации в PFN 1 гене, согласно данным полноэкзомного секвенирования (Wu et al., 2012). Расширение скрининга до дополнительных случаев БАС семейного вида позволило определить еще 3 мутации в 5 случаях семейного БАС, и p.E117G-вариант был идентифицирован на уровне очень низких частот у испытуемых контрольной группы. Дальнейший скрининг случаев БАС выявил дополнительные мутации и вариант (Ingre et al., 2013; Tiloca et al., 2013; van Blitterswijk et al., 2013; Smith et al., 2015). Затем метаанализ выявил связь варианта p.E117G с БАС, что позволило рассматривать этот вариант p.E117G как фактор риска (Fratta et al., 2014). Функция PFN1 заключается в конвертации мономерного актина в филаментный (волокнистый) актин, и также выявлено, что PFN1 локализуется в стресс-гранулах (Figley et al., 2014). Показано, что мутации PFN1 приводят к дестабилизации белка, тем самым способствуя потере функции, тогда как мутантный белок предстает в неправильно свернутом виде, что приводит к приобретению функции через искаженные белковые взаимодействия (Boopathy et al., 2015). Однако степень воздействия мутантного белка на формирование актина и динамику стресс-гранул все еще подлежит более точному установлению.
ALS22L/БАС22L: тубулин альфа 4А (TUBA4A)
Метод полноэкзомного секвенирования 363 носителей заболевания БАС семейного вида с последующим обременением редким вариантом позволил определить 5 случаев БАС с редкими вариантами в TUBA4A; они включали 4 миссенс-мутации и 1 нонсенс-мутацию, каждая из которых была закодирована в экзоне 4, в высококонсервативных аминокислотах, и эти мутации отсутствовали у 4300 контрольных участников исследования на Сервере вариантов экзом (Exome Variant Server, EVS) (Smith et al., 2014). В то время как дальнейшее секвенирование случаев БАС идентифицировало еще 1 вариант, функциональные исследования показали, что p.W407X нонсенс-мутант не локализуется в микротрубках, формируя вместо этого цитоплазматические включения, что приводит к прерыванию сборки и устойчивости микротрубок, через доминантно-негативный механизм. Последующий скрининг в китайской популяции БАС не смог идентифицировать какие-либо варианты (Li et al., 2015); данные по другим популяциям, несомненно, появятся по мере того, как опыты с использованием полногеномного экзомного секвенирования будут завершены.
Варианты промежуточного микрофиламента
Цитоскелетная дисфункция дальнейшим образом вовлекается в патогенез БАС через действие редких вариантов, которые идентифицируются в генах промежуточных микрофиламентов. Нейрофиламенты (нейроволокна) (легкие, средние и тяжелые) представляют собой основные структурные составляющие компоненты нейронального цитоскелета, и они наличествуют в характерных убиквитиновых белковых включениях. Скрининг генов-кандидатов определил редкие варианты по типу инсерции/делеции (вставки и удаления) в доменах KSP-повтора, относящихся к тяжелому гену нейрофиламента (neurofilament heavy gene, NEFH) в спорадических случаях БАС (Figlewicz et al., 1994; Tomkins et al., 1998; Al-Chalabi et al., 1999), тогда как единичная мутация «со сдвигом рамки» была идентифицирована в периферине (peripherin, PRPH1) (Gros-Louis et al., 2004). Тем не менее отсутствие мутаций в известных семейных случаях заболевания и способность показывать изолирование (сегрегацию) от болезни привели к понижению степени достоверности этих генов как локусов БАС.
Дополнительные локусы
Некоторые дополнительные локусы БАС были определены в начале 2000-х гг. Двум из них еще предстоит идентифицировать гены, которые ассоциируются с ними: ALS3 в chr18q21 и ALS7 в chr20q13 (Hand et al., 2002; Sapp et al., 2003). Еще 2 гена были определены в родословных семейного вида БАС, хотя в настоящее время прогнозируется, что их функциональное действие приводит к прерыванию нейронального развития и митохондриальной функции.
ALS19/БАС19: рецепторная тирозинкиназа 4 (ERBB4)
Полногеномное исследование японской семьи с аутосомно-доминантным БАС определило миссенс-мутацию в ERBB4 (Takahashi et al., 2013). Дополнительный скрининг установил такую же мутацию у семьи из Канады, не имеющей отношение к японской семье, и дальнейшую мутацию в БАС спорадического вида. Известно, что ERBB4 является рецепторной тирозинкиназой, которая активируется нейрегулином, что приводит к автофосфорилированию C-терминала. Мутации, происходящие в ERBB4, сокращали уровень автофосфорилирования. Было обнаружено, что ERBB4 локализуется в C-уплотнениях, образующихся из интернейронов, которые формируют синапсическую связь со спинальными двигательными нейронами (Gallart-Palau et al., 2014). Любопытен факт, что С-уплотнения не обнаруживаются в глазодвигательных нейронах, которые сохраняются при БАС, тогда как увеличение уровня нейрегулина в С-уплотнениях повышается в течение прогрессирования заболевания в модели трансгенной мыши с SOD1 G93A.
FTDALS2: суперспиральный домен-содержащий белок 10 (CHCHD10)
Изначально CHCHD10 был ассоциирован с БАС при полногеномном секвенировании семейства, выявлявшего клинические признаки, в том числе БАС (ALS), лобно-височной деменции (FTD), мозжечковой атаксии и миопатии (Bannwarth et al., 2014). Это обстоятельство привело к скринингу семейств с БАС и БАС с лобно-височной деменцией (ALS-FTD). Были выявлены некоторые дополнительные мутации (Dols-Icardo et al., 2015; Johnson et al., 2014), хотя и стало очевидно, что p.P34S-мутация была непатогенной, так как она также была обнаружена в таких же частотах у контрольных испытуемых (Marroquin et al., 2015). Функция CHCHD10 остается неизвестной; известно, что CHCHD10 локализуется в митоходрии. Фибробласты, полученые у членов семьи первоначальной родословной, показали множественные митохондриальные ДНК-делеции (удаления), нарушения респираторной цепи и структурно патологическую митохондрию, что позволило сделать вывод о том, что CHCHD10 может играть роль в респираторной цепи и (или) в устойчивости митохондриального генома (Bannwarth et al., 2014). Этот факт поддерживается дополнительным исследованием группы E.C. Genin et al. (2015), которое выявило не только потерю гребней и заживление митохондриального генома в фибробластах пациентов, но и невозможность апоптоза по причине неспособности выделять цитохром-С.
Генетика БАС оказывает существенное влияние на наше понимание заболевания и механизмов, задействованных в нейродегенерации. Большинство генов шифруют белки, участвующие в РНК-процессинге и путях деградации белков, системе убиквитин-протеасомы и аутофагии. Однако ни один из них, ни какие-либо иные задействованные пути не работают изолированно, а оказывают воздействие на другие клеточные процессы. Предполагаемые механизмы являются взаимно совместимыми, и наиболее вероятно, что многочисленные разрегулированные пути способствуют потере и гибели двигательных нейронов. Этот факт явно демонстрируется действием TDP-43, являющегося РНК-связующим белком, который неправильно локализуется из ядра, вызывая тем самым гибель ядерной функции, и скапливается в цитоплазме в качестве компонента характерных убиквитиновых включений.
Наряду с множественными генетическими причинами очевидно, что эти гены также причастны и к дополнительным расстройствам, не только к другим нейродегенеративным нарушениям, таким как лобно-височная деменция и атаксия, но и к миопатиям, костной болезни Педжета и глаукоме. Применение методов полногеномного масштабного секвенирования и полноэкзомного секвенирования в таких проектах, как 100 000 Проект геномов в Великобритании (genomicsengland.co.uk) или Project Mine Международного сообщества БАС (projectmine.com), потенциально смогут создать условия для более четкого понимания того, почему мутации гена у одного семейства представляют какой-либо конкретный клинический фенотип, тогда как другое семейство демонстрирует различные заболевания. Подобные методы исследования также продвинут наше понимание степени воздействия олигогенной наследственности при БАС.
В то время как семейные родословные явным образом демонстрируют наследственность по аутосомно-доминантному типу в классических БАС, отход от того, чтобы анализировать единичный ген в конкретное заданное время, особенно подчеркнул наличие мутаций во множественных БАС-генах, которые зарегистрированы у некоторых пациентов (van Blitterswijk et al., 2012). Скрининг крупной когорты больных БАС показал, что 14% семейного БАС и 2,6% случаев спорадического БАС имели более 1 потенциальной патогенной мутации в известном БАС-гене, и в этих случаях отмечался существенно более ранний старт развития заболевания (Cady et al., 2015). Эти данные также подчеркивают факт того, что в случаях БАС, кажущихся спорадическими, также отмечаются генетические мутации, как было засвидетельствовано при идентификации новых мутаций в случаях спорадического БАС после проведения полноэкзомного секвенирования групп из трех индивидуумов, состоящих из больных БАС и их двух незатронутых болезнью родителей (Steinberg et al., 2015; Chesi et al., 2013). Несмотря на то что в некоторых случаях эти мутации на самом деле могут представлять собой редкие мутации аутосомно-рецессивного типа, дополнительные методы WES- и WGS-секвенирования подобных случаев помогут найти объяснение тому генетическому вкладу в развитие заболевания, который был заметен в спорадических БАС и который оценивается в 61% (Al-Chalabi et al., 2010).
Существует еще одна общепризнанная теория возникновения БАС — глютаматная. Показано, что взаимодействие мутантной супероксиддисмутазы-1 с астроцитарным глютаматным переносчиком нарушает обратный захват глутамата и поэтому формируется БАС. Известно, что при БАС в области моторных нейронов наблюдается увеличение уровня концентрации глутамата (моторные нейроны, как известно, являются глутаматергическими нейронами). Увеличение глутамата не связано ни с интенсивностью нейронного повреждения, ни с распространением клинического компромисса, наблюдаемого в БАС. Это явление связывают с уменьшением главного транспортера глутамата в мозге ЕААТ2, избирательного для астроглии и служащего для очищения глутамата. Сверхстимуляция глутаматных рецепторов ведет к массивному притоку кальция в нейроны. Но глутамат в высоких концентрациях в ЦНС не ограничивается БАС, при других первичных дегенеративных нарушениях ЦНС (болезни Альцгеймера, Паркинсона, Хантингтона) также отмечаются высокие уровни глутамата в специфических областях, где нейроны находятся в стрессе (Марфунин, 2012). Формируется глутаматная эксайтотоксичность — запуск повреждения нейронов под воздействием глутамата (вещества, переносящего «информацию» в нервной системе). Однако точные механизмы, посредством которых повреждение данных генов может приводить к развитию болезни, до настоящего времени не ясны.
Глава 4. Патоморфология бокового амиотрофического склероза
Патоморфология БАС в виде повреждения передних рогов спинного мозга — это главный нозоспецифический признак этого заболевания, который впервые был описан французским исследователем Ж.-М. Шарко при аутопсии больных БАС. Более того, свое название БАС получил именно из-за очевидного при гистологическом исследовании выраженного склероза и атрофического поражения передних рогов спинного мозга (СМ) с преимущественным вовлечением в патологический процесс боковых столбов СМ, через которые проходят пирамидные тракты, начинающиеся в пояснично-крестцовом отделе СМ и заканчивающиеся в двигательной коре головного мозга (ГМ). К общим патологическим морфологическим признакам БАС относятся дегенерация кортикоспинальных трактов, связанная с потерей верхних и (или) нижних двигательных нейронов и демиелинизацией аксонов. Дегенерация кортикоспинального тракта наиболее очевидна в нижних отделах спинного мозга, но она может прослеживаться по направлению вверх через ствол мозга вплоть до уровня заднего колена внутренней капсулы и лучистого венца при помощи метода «жирового» (липидного) окрашивания, отражающего аккумуляцию макрофагов, происходящую в ответ на дегенерацию миелина (Rowland et al., 2001). Этот феномен преимущественного повреждения двигательных нейронов в передних рогах спинного мозга при БАС также в конце XX в. определил новое название этой болезни во всем мире — «болезнь моторных нейронов». Сегодня морфологи во всем мире доказали, что патологическим изменениям при БАС подвергаются не только двигательные нейроны, но и почти все клетки и элементы (синапсы, волокна, межклеточные контакты и т.д.) нервной ткани, вовлеченные в патологический процесс. Более того, показано, что и не все мотонейроны страдают при БАС, а только определенная часть из них. Часть нейронов черепно-мозговых нервов остается интактна к повреждающим этиопатогенетическим факторам. Это говорит о том, что двигательный нейрон не является специфической мишенью этиопатогенетических факторов этой болезни. Просто из-за своих больших размеров и особых функций по передаче пакетов электрических импульсов они более других клеток страдают от эксайтотоксического воздействия аутореактивных лимфоцитарных клеток на клеточные системы в нервной ткани, вовлеченные в асептический воспалительный процесс. Наиболее вероятно, что при боковом амиотрофическом склерозе дегенерация мотонейрона — не «начало», а «конечный путь» каскада общепатологических патофизиологических реакций, инициируемых различными известными или неизвестными триггерами, которые завершаются грубым морфологическим дефектом мотонейронов. Исходом этого каскада является морфологическая системная нейродегенерация клеток вовлеченной нервной ткани с преимущественным повреждением и атрофией двигательных проводящих путей. Как мы уже отмечали, в некоторых случаях БАС может быть обусловлен мутациями в гене супероксиддимутазы-1, когда основным патогенетическим фактором служит цитотоксическое действие дефектного фермента. Мутантная супероксиддисмутаза-1 накапливается меж слоев митохондриальной мембраны, нарушая аксональный транспорт, и взаимодействует с другими белками, что приводит к нарушению их деградации. Возникновению спорадических случаев БАС якобы способствуют неизвестные триггеры, которые так же, как и мутантная супероксиддисмутаза-1, способны к реализации своих эффектов в условиях повышенной функциональной нагрузки на мотонейроны, что вызывает их селективную уязвимость. В результате усиления функций мотонейронов повышается уровень выброса глутамата, накапливается избыток внутриклеточного кальция, активируются внутриклеточные протеолитические ферменты, а выделяемый из митохондрий избыток свободных радикалов повреждает клетки микроглии, астроглии, а также сами мотонейроны с их последующей дегенерацией. Это стандартный механизм, объясняющий появление морфологического дефекта в нервной ткани при БАС. То есть БАС рассматривается в современных учебниках и руководствах по неврологии как истинное заболевание моторного нейрона. Отсюда и появление нового названия болезни — БМН, которое акцентирует проблему и концентрирует ее на патологии отдельных мотонейронов. С одной стороны, получается, что на двигательный мотонейрон якобы направлено все этиопатогенетическое воздействие болезни и он является причиной и следствием воздействия всего каскада нейропатологических изменений, возникающих в мотонейроне. С другой стороны, нейропатологические подходы к нейродегенеративным расстройствам подвергались критике по причине того, что некоторые нейроученые считают, что дегенерирующие нейроны лишь представляют собой патологии «конечной стадии». Paul G. Ince (2000) считает, что есть две причины, согласно которым этот взгляд интерпретируется неверно. Во-первых, больные с нейродегенеративными расстройствами, в том числе БАС, погибают в результате случайных причин или сопутствующих заболеваний (например, ишемической болезни сердца) так же, как и представители общей популяции. Любое крупное собрание тканей больных БАС, полученное при аутопсии, будет включать тех больных, которые умерли неожиданно в силу разнообразных причин на ранней стадии болезни. Во-вторых, степень распространения патологии БАС по ЦНС варьируется в каждом индивидуальном случае. Многие больные на момент летального исхода обнаруживают тяжелую форму бульбарного шейно-торакального заболевания с относительным сохранением поясничного отдела спинного мозга. В подобных случаях представляется возможным провести сравнение тяжело пораженных и умеренно пораженных участков двигательной системы у одного и того же больного. Наблюдения за больными, умершими неожиданно на ранней стадии болезни, при сравнении тяжело пораженных участков с относительно сохраненными группами двигательных нейронов нижних конечностей, наблюдаемыми у некоторых больных, не смогли выявить какую-либо последовательную «раннюю» патологию, которая не замечалась бы на более поздних стадиях заболевания.
При стандартном патоморфологическом исследовании БАС находят следующие общие объективные феномены морфологических изменений:
• избирательная атрофия передних двигательных корешков и клеток передних рогов спинного мозга; наиболее выраженные изменения происходят в шейных и поясничных сегментах, но при этом задние чувствительные корешки остаются нормальными;
• в нервных волокнах боковых кортикоспинальных трактов спинного мозга наблюдается демиелинизация, неравномерное набухание с последующим распадом и гибелью осевых цилиндров, что обычно распространяется и на периферические нервы;
• в некоторых случаях отмечается атрофия прецентральной извилины большого мозга, иногда атрофия захватывает VIII, X и XII пары черепных нервов, наиболее выраженные изменения происходят в ядре подъязычного нерва;
• атрофия или отсутствие мотонейронов, сопровождающиеся умеренным глиозом без признаков воспаления;
• утрата гигантских пирамидных клеток (клетки Беца) двигательной коры;
• дегенерация боковых пирамидных путей спинного мозга;
• атрофия групп мышечных волокон (в составе двигательных единиц).
На рисунке 5 представлены фотографии моторных нейронов в норме (здоровый мотонейрон), а также формирование дегенерации мотонейрона при боковом амиотрофическом склерозе в фазе лейкоцитарной инфильтрации и последующей дегенерации двигательного нейрона. Очевидно, что инфильтрация мотонейрона лейкоцитами является наиболее очевидным фактором формирования дегенеративного процесса, в результате которого в нем происходит мощнейший оксидативный стресс и запускаются основные молекулярно-биологические механизмы патогенеза.
Одной из причин возникновения лейкоцитарной инфильтрации вокруг моторных нейронов является нарушение гемато-энцефалического барьера (ГЭБ) при БАС. Считается, что нарушение ГЭБ при БАС вызвано всем спектром этиопатогенетических факторов этого заболевания, в котором значимую роль играют нарушения сосудистой проницаемости вовлеченных в патологический процесс микрососудов головного и спинного мозга, обусловленные асептическим воспалением, реактивностью микроглии и астроцитов и значительным цитокиновым взрывом. В целом иммунологическая дисфункция и системное воспаление являются основными факторами в нарушении целостности ГЭБ.
Рис. 5. Фотографии моторного нейрона в норме (1), при лейкоцитарной инфильтрации моторного нейрона с формированием зоны асептического воспаления (2) и при выраженной дегенерации при боковом амиотрофическом склерозе (3)
Примечательно, что усиленная адгезия и миграция лейкоцитов через ГЭБ формируют некоторые фундаментальные звенья патогенеза, при этом наибольшее внимание уделяют изучению роли тучных клеток в стресс-индуцированном повышении проницаемости ГЭБ, что способствует развитию нейровоспаления, активации и повреждению клеток. В числе механизмов, запускающих такие процессы, — ответ на поврежденные клеточные макромолекулы (DAMPs), например митохондриальные ДНК, секретируемые во внеклеточное пространство в очаге иммунного воспаления и обладающие нейротоксическим действием, проявляющимся поведенческими изменениями.
Воспалительный процесс, затрагивающий микроциркуляторное русло, вызывает активацию глиальных клеток и секрецию медиаторов воспаления в клетках головного и спинного мозга. При этом на эндотелиоцитах увеличивается экспрессия молекул адгезии, что способствует проникновению лейкоцитов в мозговую ткань. Например, увеличение экспрессии эндотелиоцитами ИЛ-6 и PGE2 предполагает, что эти медиаторы участвуют в передаче воспалительного сигнала на другие клетки головного мозга (астроциты, микроглия, перициты и периваскулярные макрофаги), что проявляется при патоморфологическом исследовании нервной ткани у больных с БАС. Помимо этого, при прогрессировании дегенеративных заболеваний наблюдается увеличение пассивного транспорта веществ через открывшиеся плотные контакты, повышение везикулярного транспорта.
Все вышеописанные системные морфологические изменения в нервной ткани головного и спинного мозга при БАС хорошо известны и практически не вызывают сомнений у нейроспециалистов. Они были очень подробно описаны еще в конце XIX в. и очень тщательно уточнены в середине и конце XX в. целой плеядой русских и зарубежных неврологов (Ходрариан, 1957). В настоящее время установленные ранее характерные анатомо-морфологические проявления касаются преимущественно классического БАС и убедительно подтверждены гистологическими, гистохимическими и иммуногистохимическими данными. Но в последнее время появилась целая серия новых научных фактов об атипичной морфологии БАС. Они стали значительно расширяться за счет появления большого количества структурно-морфологических данных при аутопсии нейродегенеративных заболеваний, очень похожих на БАС. Группа схожих между собой и родственных расстройств двигательной системы (ALS-деменция, прогрессирующая мышечная атрофия, первичный боковой склероз и «болезнь двигательного нейрона с включением деменции» — motor neuron disease, MND inclusion dementia) в данном контексте рассматриваются в свете подобных патологических проявлений, присущих БАС. Все они могут встраиваться в концептуальные рамки, характеризующие общую молекулярную морфологическую патологию, которая предстает с существенным фенотипическим многообразием, на основе анатомической вариабельности в степени индивидуальной восприимчивости к патологии. Взаимосвязь между подобными патологическими признаками, выявляемыми при аутопсии, а также нынешний взгляд на морфогенез заболевания рассматриваются с учетом постановки ряда связанных друг с другом вопросов отдельными зарубежными нейроспециалистами (Ince, 2000):
a) Характеризует ли единичная специфичная молекулярная патология заболевание БАС как самостоятельную нозологию, как, например, это происходит при сенильных бляшках и нейрофибриллярных клубках при болезни Альцгеймераили с тельцами Леви, характерными для болезни Паркинсона?
b) Какова патоморфологическая взаимосвязь между семейным и спорадическим видами БАС?
c) Характеризуют ли схожие молекулярные патологии какой-либо конкретный БАС, обусловленный мутациями в гене, кодирующим супероксиддисмутазу медь-цинка (SOD1) и другие формы спорадического и семейного БАС?
d) Есть ли морфологические различия между другими генетически детерминированными дегенерациями двигательного нейрона (например, связанная с X- хромосомой бульбоспинальная мышечная атрофия Кеннеди, спинальная мышечная атрофия) и разнообразными формами БАС?
Отсутствие in vivo методов мониторинга за патологическими изменениями в двигательных нейронах и их аксонах привело к тому, что представление о процессе заболевания базируется более на перекрестных (кросс-секционных) данных, нежели на данных продольных (лонгитудинальных) наблюдений. Таким образом, ключевые вопросы, касающиеся механизмов клеточной гибели, а также кинетики процесса, продолжают оставаться спорными применительно к заболеванию человека, несмотря на то что доступны дополнительные данные, полученные из in vivo исследований морфологических изменений в двигательных нервах. Схожие проблемы возникают при интерпретации изменений, происходящих в верхних двигательных нейронах при БАС. Два клинических подхода, призванных усовершенствовать наше представление о дегенерации двигательной системы при БАС, состоят из различных in vivo режимов визуализации (структурной и функциональной магнитно-резонансной визуализации и магнитно-резонансной спектроскопии) и более современных электрофизиологических подходов (например, транскраниальной магнитной стимуляции, воздействующей на двигательный путь). Представления о патогенезе и патоморфологии БАС будут продолжать видоизменяться по мере появления новых данных, получаемых при изучении молекулярной патологии, клинико-патологических исследований и работ с трансгенными моделями дегенерации двигательной системы, наряду с данными, получаемыми от in vivo методов по работе с человеком.
Типичная клиническая картина комбинированных признаков расстройства верхних двигательных нейронов и нижних двигательных нейронов является преобладающей при классических формах БАС, и она отображается в анатомической дистрибуции дегенеративных изменений в двигательной системе этих больных (Ходкариан, 1957). До настоящего времени классическое морфологическое описание морфологического повреждения при БАС сильно не изменилось. Преобладающие патологические характеристики, коррелирующие с основными клиническими проявлениями, сводятся к следующему:
a) сокращение как количества, так и размера нижних двигательных нейронов (клеток переднего рога) в передних рогах серого вещества спинного мозга и бульбарных двигательных ядрах;
b) миелиновое побледнение в кортикоспинальном проекционном проводящем пути, что является вторичным следствием аксональной гибели в этом участке.
Помимо общих или системных патоморфологических изменений, характерных для БАС как нозологической единицы нервных болезней человека, морфологи выделяют локальные морфологические изменения, характерные для конкретных топографических отделов ЦНС. То есть локальные морфологические поражения отдельных нейронов также имеют определенную специфику и хорошо описаны гистологами в научной литературе. Патологии, происходящие в теле верхних двигательных нейронов (клетках Беца), намного более разнообразны, и с помощью традиционных и иммуноцитохимических окрашиваний были описаны многие дополнительные патологические особенности. Остановимся более подробно на типичных морфологических изменениях в нервной ткани головного и спинного мозга больных БАС, характеризующих повреждение и дисфункцию как непосредственно в двигательной сфере организма, так и вне двигательной сферы.
Верхние моторные нейроны двигательной коры головного мозга. Степень, до пределов которой патологические изменения выражены при аутопсии двигательной коры, носит весьма переменный характер даже в случаях с клинически типическими признаками дисфункции верхних двигательных нейронов. Случаи наиболее тяжелого поражения демонстрируют очевидное отсутствие гигантских клеток Беца в 5-м кортикальном слое во 2-й степени, что сопровождается реактивным глиозом, предстающим в виде астроцитоза, который, например, можно доказать при помощи иммуноцитохимии глиального фибриллярного кислотного белка (GFAP), а также в виде диффузного микроглиоза, который верифицируется с использованием иммуноцитохимии.
В большинстве случаев в двигательной коре головного мозга наблюдается отмирание клеток Беца, а в лобных и теменных долях происходит атрофия, что подтверждается исследованием МРТ при первичном БАС (Живолупов и др., 2011). Происходит опустошение других волокон, расположенных в вентральных и боковых канатиках, что вызывает характерную бледность окрашенного миелина. Однако данное состояние может обуславливаться и утратой коллатералей двигательных нейронов, которые способствуют lamina propria (собственной пластинке). Такой же эффект можно наблюдать и при долгосрочной, тяжелой форме полиомиелита (Maurice, Allan, 2001). Крупные нейроны подвергаются негативному воздействию раньше, чем небольшие. Утраченные клетки замещаются фиброзными (волокнистыми) астроцитами. Многие из выживающих нейронов имеют небольшой размер, сжатую форму и заполняются липофусцином. Периодически можно наблюдать аномально измененное цитоплазмическое тельце-включение. Согласно некоторым сообщениям, вздутие (отек) проксимального аксона представляет собой ранний признак, предположительно предшествующий видимым изменениям, происходящим в самом теле клетки. Передние корешки сужены в размере, и в двигательных нервах наблюдается непропорциональная потеря крупных миелинирующих волокон. Мышцы показывают типичную денервационную атрофию различного масштаба. Группа Whitehouse с коллегами выявила истощение и опустошение мускариновых, холинергических, глицинергических и бензодиазепиновых рецепторов в тех областях спинного мозга, откуда исчезли двигательные нейроны (Whitehouse et al., 1983).
К сожалению, подобные результаты не являются прямым доказательством того, что клетки Беца исчезают на самом деле, так как не существует молекулярного маркера, который бы исключительно маркировал верхние двигательные нейроны. С фенотипической точки зрения эти крупные глутаматергические нейроны обладают молекулярными характеристиками, которые схожи с другими пирамидальными клетками коры мозга. То, насколько явная гибель клеток Беца может быть замечена с помощью традиционных методов (окрашивания по Нисслю), может частично обуславливаться нейрональным сокращением, приводящим к тому, что клетки Беца становятся неотличимыми на основе лишь одних критериев размера от соседних пирамидальных клеток, что, вероятно, вариативно в каждом конкретном случае. Дополнительный фактор, подлежащий рассмотрению при изучении индивидуальных случаев, состоит в длительности и степени тяжести проявлений патологии верхних двигательных нейронов. Как показывает опыт P.G. Ince (2000), некоторые случаи БАС с ясными признаками дисфункции верхних двигательных нейронов на протяжении нескольких лет могут демонстрировать удивительно сохранные и практически нормальные морфологические проявления в двигательной коре головного мозга.
Внутриклеточные поражения являются чрезвычайно редкими в уцелевших клетках Беца у больных с типичным спорадическим БАС, если только они не сопровождаются деменцией (слабоумием) или связаны с мутацией в SOD1-гене. Литературные источники содержат противоречащие сведения об астроцитарном глиозе, который отмечается в некоторых случаях БАС. Согласно опыту P.G. Ince (2000), наиболее распространенной формой является конфлюэнтный (сливающийся) диффузный глиоз, попеременно распространяющийся по направлению вверх от участка соединения белого и серого веществ и соединяющийся с интенсивным субкортикальным глиозом. Другие описания, свидетельствующие о более очаговом и фрагментарном глиозе (Kamo et al., 1987), могут отражать гиперболизацию и преувеличение здорового (обычного) прерывистого паттерна кортикального астроцитарного окрашивания на глиальный фибриллярный кислый протеин (GFAP).
Верхние моторные нейроны пирамидного тракта. В большинстве случаев БАС обнаруживаются доказательства побледнения миелина кортикоспинального тракта на каком-либо уровне этого проводящего пути. Подобные изменения следует интерпретировать в свете известных анатомических особенностей этого пути, встречаемых у людей. Во-первых, размер проекции, с учетом количества проецируемых аксонов, указывает, что клетки Беца влияют лишь на незначительную долю от общей численности. Данный факт подразумевает, что многие пирамидные клетки, расположенные в моторном участке и, по всей видимости, в премоторной области (поля Бродмана 4 и 6), способствуют прямой кортикоспинальной проекции в ствол головного мозга и спинной мозг (Chou, 1995). Во-вторых, плотность терминальной иннервации пути в спинном мозге носит переменчивый характер. Работы, изучающие высших приматов, дают основания предположить, что большинство моторных волокон проецируют по направлению к нижним двигательным нейронам и интернейронам, расположенным в цервикальном утолщении вокруг периферии переднего (вентрального) рога. Эти анатомические данные отражают основное функциональное значение этого пути при полном двигательном контроле верхних конечностей. Как следствие, размер кортикоспинальных трактов, располагающихся ниже первого торакального спинального уровня, намного снижен по сравнению с медуллярной или цервикальной областями. Степень перекрещивания в спинномозговом веществе является изменчивой как относительно пропорции пересеченных аксонов, так и применительно к симметрии перекрещивания.
Ранее исследовательская группа Brownnell (1970) обратила внимание на то, что побледнение кортикоспинального тракта в миелиновых окрашиваниях наиболее всего заметно в цервикальном отделе мозга и медуллярных пирамидах, что подтверждено, по опыту авторов, более 90 случаями. В некоторых случаях подобные явления очевидны на всех исследуемых уровнях, так что передняя конечность внутренней капсулы, центральная церебральная ножка и канатики белого вещества бокового и вентрального спинального отделов подвержены негативному воздействию. Преобладание случаев, в которых миелиновое побледнение не отмечается над уровнем мозгового вещества спинного мозга, включая случаи с неявной патологией двигательного кортекса, приводит исследователей к выводу о том, что патологические изменения в верхних моторных нейронах при БАС чаще всего происходят по причине аксонопатии, сопровождаемой периферическим «отмиранием».
Известный с конца прошлого века опыт, полученный при использовании маркеров для макрофагов (Ince et al., 1996; Ince et al., 1998), позволяет предположить, что метод иммунного окрашивания с CD68 (или иными маркерами), возможно, является более чувствительным методом выявления изменений кортикоспинального тракта при отсутствии четкого доказательства миелинового побледнения. Подобные наблюдения требуют осторожности, учитывая степень, до которой бесчисленные поражения мозга, в том числе гипоксия, ассоциируются с повышенной активностью макрофагов и микроглии. Подтверждение неявной кортикоспинальной патологии следует интерпретировать лишь в случаях с типичным во всех иных отношениях молекулярным патологическим доказательством наличия БАС или болезни моторного нейрона (БМН), когда паттерн микроглиального окрашивания сосредоточен в участках белого вещества, о которых известно, что они подвергаются негативному воздействию при БАС, и при отсутствии подтверждения причины диффузной «неспецифической» микроглиальной активации. Включение подобных случаев, вероятно, сможет улучшить клинико-патологическую корреляцию между признаками расстройства верхних моторных нейронов и «патологией верхних моторных нейронов».
Нижние моторные (двигательные) нейроны. Гибель нижних моторных нейронов при БАС часто субъективно более выражена и заметна, чем это может быть верифицировано при помощи методов нейронального подсчета. Вероятно, две основные причины лежат в основе этого научного факта. Во-первых, сам механизм нейрональной дегенерации при БАС, по-видимому, происходит преимущественно за счет сокращения объема нейрона, так что тщательное исследование способно выявить пропорцию уцелевших небольших темных моторных нейронов (Hiranj, Iwata, 1978). Ранее также было показано, что эти нейроны демонстрируют многие биохимические свойства «программированной клеточной гибели», или апоптоза (Martin, 1999). Во-вторых, наблюдения на отдельных участках на любом спинальном уровне являются недостоверными из-за большой вариативности количества тел в общей совокупности моторных нейронов, присутствующих между одним участком и следующим за ним (Tomlinson et al., 1997). Вследствие этих причин наличие характерных спинальных (или бульбарных) телец включений моторных нейронов является убедительной составляющей общей патологической картины. Тем не менее аспекты подобной гибели моторных нейронов представляют интерес в понимании механизмов и динамики моторной нейрональной дегенерации.
Вероятность того, что гибель нейронов при нейродегенеративных заболеваниях может вызываться через механизм «программируемой гибели клеток», известный как апоптоз, вызывает стабильный интерес у ученых (Martin, 1999). Но здесь мы приведем только несколько комментариев к этому термину. Во-первых, вся концепция клеточной гибели часто представлена в виде выбора «один-или-другой» между некрозом и апоптозом, но подобная концепция не может применяться в отношении взрослой ЦНС. Показано, что глутаматергическая нейротоксичность может обладать морфологическими характеристиками в континууме некроза-апоптоза, в том числе «промежуточными формами», наделенными свойствами обоих режимов клеточной гибели (Portera-Cailliau et al., 1995). Во-вторых, в соответствии с общим опытом нейропатологов, исследовавших случаи БАС, классические апоптозные тела (подобные тем, которые рутинным образом встречаются в образцах опухоли мозга) не видимы. Напротив, наличие небольших сжатых моторных нейронов широко описывается в патологических отчетах по БАС, и они являются намного более вероятным кандидатом для морфологической «предыстории» итоговой или окончательной нейрональной гибели. Дальнейшее подтверждение возникновения нейронального сжатия, которое происходит до момента нейрональной гибели, подкрепляется работами с периферическими нервами: количественный анализ выявил систематическое сокращение аксональных диаметров, сжимающихся от крупных до средних калибров, что является более значимым и выраженным, чем общее падение абсолютной численности аксонов в нейронах моторных нервов при БАС. Данное наблюдение коррелирует с изменениями в вентральном (переднем) роге и подразумевает, что по мере того, как общая совокупность нейрональных телец (neuronal somata) сжимается, диаметры их аксонов также пропорционально сокращаются («аксодендритическое обрезание») (Martin, 1999).
Большинство исследований, пытавшихся количественно подсчитать утрату нижних моторных нейронов в поясничном утолщении, показывают среднюю потерю в 50% по сравнению с контрольными участниками сопоставимого возраста (Ince et al., 1995). Наиболее всеобъемлющие данные были собраны по 11 случаям, изучаемым группой B.E. Tomlinson и коллегами (данные были опубликованы в монографии Amyotrophic Lateral Sclerosis под редакцией R.H. Brown, V. Meininger, M. Swash, 2000). Эти случаи были подобраны в соответствии с локализацией патологического процесса в пояснично-крестцовом утолщении, используя 20 мкм срезы, которые в дальнейшем были подсчитаны из расчета на каждые 100 мкм (например, каждый пятый срез). Результаты представлены по оценкам общего количества нижних моторных нейронов по спинальному уровню L4, в сравнении с данными, полученными тем же методом по 65 контрольным случаям, в возрасте между 25 и 95 годами.
Здесь важно отметить следующее. Во-первых, данные по контрольным участникам изначально были опубликованы с анализом, указывающим, что нейрональное выпадение при естественном старении не начиналось до 65-летнего возраста (Tomlinson et al., 1977). Повторный анализ этого массива данных, выполненный с помощью компьютеризированного математического моделирования, показывает, что данные недостаточно убедительны, чтобы допускать разграничения в любой функции, задействованной для того, чтобы соответствовать данным. Таким образом, изначальная двухфазовая интерпретация обладает не большей достоверностью, чем, например, простая линейная модель, где нейрональная гибель начинается в третьем десятилетии жизненного цикла (Ince, Appleton, наблюдения не опубликованы). По этой причине контрольный массив данных показан с 95-процентными доверительными интервалами, с использованием метода простого линейного приближения к данным. Во-вторых, данные по БАС показывают очевидную обратно пропорциональную зависимость от возраста. Эти данные не достигают статистической значимости на уровне 95%, но могут указывать на то, что более молодые больные имеют тенденцию к более длительной выживаемости, что сопровождается пропорционально более высокой нейрональной гибелью, что, в свою очередь, обусловлено более высоким общим уровнем «приспособленности» на момент начала заболевания. Тот факт, что у некоторых больных сохраняется численность нейронов, сопоставимая с нормальными значениями, также иллюстрирует то, насколько переменна и изменчива может быть местная региональная вовлеченность в патологический процесс всего спинного мозга. Больные могут умереть от респираторной дисфункции в результате бульбарной или цервикально-торакальной патологии на той стадии, когда они еще могут передвигаться и поражены минимальной пояснично-крестцовой слабостью. Напротив, некоторые больные предстают с признаками патологии нижних конечностей, и у них развивается центростремительный паттерн спинальной вовлеченности, при котором рано развивается паралич нижних конечностей. При аутопсии региональная интенсивность гибели нейронов коррелирует с паттерном клинического заболевания, однако разграничений в отношении клеточной или молекулярной патологии у больных в пределах данного спектра не существует.
По-видимому, нет разницы в восприимчивости моторных нейронов и конечностей по сравнению с более медиально расположенными аксиальными моторными нейронами вентральных (передних) рогов. Однако две группы типичных во всех иных отношениях моторных нейронов характерным образом сохраняются в большинстве случаев:
a) крестцовое моторное ядро Онуфа-Онуфровича (ядро Онуфа), которое иннервирует мышцы тазового дна и причастные к ним сфинктеры (Ivata, Hirano, 1978); подобное патологическое сохранение коррелирует с задержкой мочевой и фекальных функций как с типичным клиническим проявлением на всем протяжении заболевания; и
b) моторные нейроны 3-й, 4-й и 6-й пар черепно-мозговых нервов, иннервирующих внешние окулярные мышцы, которые также характерным образом сохраняются. Это приводит к задержке здорового движения глаз, которая также является типичным признаком заболевания.
Основа подобного избирательного сохранения, вероятно, состоит в сложных взаимодействиях физиологических и биологических свойств этих нейрональных групп, которые имеют отношение к некоторым возможным механизмам заболевания. Отдельный интерес могут представлять (Alexianu et al., 1994; Ince et al., 1993):
a) экспрессия цитоплазматических кальциевых буферных белков в сохраненных моторных ядрах;
b) недостаточное количество прямой моносинаптической кортикоспинальной иннервации;
c) различия в глутаматергической нейротрансмиссии;
d) метаболизм окиси азота.
При БАС также отмечается немоторная патология ЦНС, и представлять болезнь как исключительное повреждение мотонейронов неверно. Вовлеченность экстрамоторных участков ЦНС в патологический процесс, обусловленный БАС, подтверждается при аутопсии многих больных БАС. Клинические корреляты чувствительных, церебеллярных и экстрапирамидных свойств редко описываются у этих больных, но более чувствительные объективные измерения (например, вызванные потенциалы, формальная психометрия, функциональная визуализация мозга) обычно демонстрируют умеренные нарушения, выявляемые при помощи подобных методов диагностики.
Чувствительная (сенсорная) система. Бледность дорсальных канатиков спинного мозга — достаточно частое явление при БАС, хотя она особенно подчеркивается в семейных случаях, когда выявляется у 70% больных (Ivata, Hirano, 1997; Lawyer, 1953). Эту бледность можно наблюдать в миелин-окрашенных участках, и она обычно преобладает в «зоне входа дорсального корешка», расположенной в клиновидном пучке. Кроме того, бледность часто отмечается в тонком пучке (Ivata, Hirano, 1997). Несмотря на то что подобные изменения ассоциируются с семейным заболеванием, они также отмечаются и в спорадических случаях (Hadson, 1981). Детальные исследования, которые сообщали бы об относительной частоте данной патологии в семейном и спорадическом видах заболевания, отсутствуют. Такие исследования особенно трудновыполнимы при подобном заболевании, сопровождаемом поздним возникновением и быстрым летальным исходом. В сериях случаев крупных «современных» аутопсий о вовлечении нейронов в спинальных задних рогах спинного мозга не сообщается. Тем не менее такое вовлечение, несомненно, отмечается в некоторых семейных случаях, связанных с мутациями, происходящими в гене, который кодирует SOD1. Кроме того, хорошо задокументирована нейрональная атрофия в ганглиях задних корешков спинномозговых нервов (posterior root ganglia), периферических чувствительных нервах и таламический глиоз (Brounell et al., 1970).
Церебеллярные (мозжечковые) пути. Некоторые работы также часто отмечали изменчивую бледность миелина в восходящих спинно-церебеллярных путях и в торакальном ядре Кларка, которое дает начало дорсальному спинно-церебеллярному пути (Swash et al., 1986). В типичных случаях БАС отмечается диффузное побледнение всего бокового и вентрального белого вещества, в том числе в этих церебеллярных проекциях. Эти изменения интерпертируются как доказательство вовлечения «косвенных» нисходящих кортикоспинальных моторных путей в патологический процесс. Нижние ядра олив могут быть подвержены транснейронной дегенерации, но доказательств того, что мозжечковые нейроны уязвимы при БАС, нет. Подобным образом проекционные зоны церебеллярного продуцирования (например, вентральные мостовые ядра, относящиеся к варолиевому мосту), не подвержены негативному воздействию при БАС.
Черная субстанция. Существует очень небольшое количество сообщений о сочетании БАС с «идиопатической болезнью телец Леви и болезни Паркинсона» (Eisen, Calne, 1992). В проделанном P.G. Ince (2000) исследовании 90 случаев БАС, верифицированных при аутопсии, есть 2 больных, у которых дегенерация черного вещества была связана с тельцами Леви, несмотря на то что клинический диагноз болезни Паркинсона был поставлен только у одного из них (Wiliams et al., 1995). Чтобы вынести комбинированный диагноз, необходимо выявить как типичные анатомические особенности, так и молекулярные патологические характеристики обоих расстройств. Однако менее тяжелая форма дегенерации черного вещества, так называемый субклинический паркинсонизм, отмечается у многих больных с БАС. В этих случаях он приобретает форму дегенерации дофаминергических нейронов черного вещества по характерной дегенерации для телец не-Леви. В тех случаях, когда подобные изменения удалось количественно подсчитать при аутопсии, наблюдалась взаимосвязь между выраженной дегенерацией черной субстанции и больными, у которых БАС комбинировался с синдромом фронтальной лобной деменции (Burrow, Blumberg, 1992). Дополнительные данные, полученные у больных БАС, свидетельствующие в пользу сокращения размера дофаминергической проекции до уровня базальной ганглии, получены в результате использования методов in vivo сканирования с использованием 6-флуородопа, который отображает серьезные изменения примерно у одной четверти обследованных больных. Механизм, через который осуществляется гибель клеток черной субстанции при отсутствии специфических молекулярных патологических изменений, пока не установлен.
Отдельного обсуждения требует патология, не связанная с деятельностью ЦНС (Non-CNS pathology), и эти морфологические проявления болезни мы обсудим ниже.
Мышцы. Типичные изменения, происходящие при атрофии скелетной мускулатуры, описаны в самых ранних сведениях о патологии БАС (Aran,1850; Charcot, Jofroy, 1869; Kojevnikoff, 1883; Martin, Swash, 1995). Эти изменения заключают в себе особенности денервационной атрофии с кластерами ангулярных атрофических волокон. Это кластерообразное скопление, наряду с группированием по волоконному типу в атрофических кластерах, представляет собой неспецифическое следствие серийного последовательного цикла денервация — реиннервация — денервация. При БАС реиннервация берет свое начало из коллатерального (побочного) спраутинга внутримышечных аксонов (Wohlfart, 1957).
Другие ткани. При БАС неспецифические патологические изменения были заявлены как выходящие за пределы ЦНС. Например, в коже отмечается перемена в коллагеновом перекрестном связывании или в пропорции незрелого коллагена наряду с более быстрой дегенерацией в компоненте эластина (Ono, Yamauchi, 1992). Вопрос о том, обладают ли подобные изменения какими-либо клиническими коррелятами (например, с относительной редкостью (нерегулярностью) циркуляторных варикозных язв при БАС), подлежит обсуждению. Отмечались коллагеновые патологии при аутопсии спинного мозга больных БАС по сравнению как с контрольными группами здоровых участников, так и с участниками, переносящими нейродегенеративные расстройства (Ono, Yamauchi, 1994). Сообщения о разбухании митохондрий и внутриклеточных включениях в печени при БАС не вполне достоверны.
Морфологические особенности деменции при БАС. В настоящее время хорошо известен факт, что до 5% больных с типичными моторными признаками, характерными для БАС, манифестируют в виде синдрома лобно-височной деменции, либо незадолго до, либо вскоре после наступления моторного расстройства. Церебральная патология данного синдрома объединяет несколько неспецифических признаков. Наблюдается диффузная атрофия церебральных полушарий, сопровождаемая акцентировкой в лобно-височном отделе (Kew, Leigh, 1992), наряду с микровакуолизацией кортикального слоя 2 в наиболее сильно пострадавших участках (Lowe, 1994). Еще одним неспецифическим признаком является наличие переменного, но обычно интенсивного субкортикального глиоза белого вещества в лобно-височных областях; этот глиоз может расширяться до уровня хвостатого ядра (Kew, Leigh, 1992). Недавно проделанное стереологическое исследование указало, что диффузная кортикальная нейрональная гибель может иметь широко-распространенный характер даже у больных БАС при отсутствии деменции (Latsoudis et al., 1999). Дегенерация черного вещества при отсутствии формирования тел Леви является еще одним общим признаком.
Наиболее специфической чертой патологии данной категории больных считается более широкое распространение наличия тел убиквитинированного нейронального включения, чем при типичном БАС (Wighman et al., 1992; Neary et al., 1990; Peavy et al., 1992). Они характерно присутствуют в гиппокампальных гранулярных клетках зубчатой извилины и, в более переменном виде, в нейронах 2-го и 3-го кортикальных слоев лобно-височных отделов. Исследования молекулярной патологии указывают, что убиквитинированные нейрональные тела включения разделяют те же признаки, что и убиквитинированные включения (UBIs) моторных нейронов, наблюдаемые при БАС. Тем не менее, так как основная белковая конституция убиквитиновых включений (UBIs) еще не была показана, эта интерпретация в некоторой степени носит гипотетический характер. С концептуальной точки зрения предполагается, что у некоторых больных восприимчивость к процессу заболевания, характеризующему БАС, распространяется до церебральных участков, которые обычно не подвергаются патологическому воздействию. Подобная церебральная дегенерация проявляется патологически через наличие UBIs (убиквитинированных включений) в церебральных кортикальных участках, а дистрибуция патологии создает условия для адекватного анатомического субстрата, необходимого для формирования нейропсихологических признаков БАС-деменции. Дальнейшее подтверждение подобной концепции о расширенном региональном вовлечении в патологический процесс при БАС, которое простирается до уровня немоторных зон, происходит из наблюдений за больными с БАС и без наличия деменции. Исследования церебральной функции, выполненные при помощи позитронно-эмиссионной томографии (Ladolph et al., 1992), а также подробное нейропсихологическое тестирование больных БАС без наличия деменции (Chari et al., 1996) показывают, что у многих больных выявляются доказательства субклинического церебрального вовлечения немоторных зон. Кроме того, патологическое обследование гиппокампа пациентов с БАС при аутопсии показывает наличие гиппокампальных включений, отмечаемых у большего количества случаев, чем о них было известно как о сигнализирующих о когнитивных нарушениях в жизни (Lowe, 2000).
Известные морфологические варианты БАС. Традиционный взгляд на БМН (болезнь моторного нейрона) рассматривает 3 синдрома: синдром классического БАС, синдром прогрессирующего бульбарного паралича и синдром прогрессирующей мышечной атрофии как варианты одного и того же процесса заболевания (Dejerin, 1883). Большинство случаев, которые предстают в качестве прогрессирующего бульбарного паралича, продолжают развиваться и приобретают форму типичного БАС на протяжении болезни. Больше разногласий сохраняется по поводу статуса первичного БАС применительно к данному спектру заболевания. Современные патологические исследования, основывающиеся на определении процессов заболевания через использование молекулярных патологических маркеров, указывают на то, что как первичный БАС, так и тип лобно-височной деменции (деменция включения болезни моторного нейрона — MND inclusion dementia) должны рассматриваться как синдромы, возникающие через тот же патологический каскад, что и при БАС-болезни моторного нейрона (ALS-MND) (Jakson et al., 1996), но с различной анатомической избирательностью.
Патологические наблюдения, которые связывают эти расстройства, приводятся ниже. Ключевым элементом, позволяющим четко установить эти синдромы внутри целого спектра нейродегенерации, являются убиквитинированные включения (UBI). В настоящее время морфологические (полученные при световой и электронной микроскопии) патологические результаты и молекулярные патологические результаты позволяют предположить, что UBI представляют собой характерное для БАС повреждение, несмотря на несовершенное понимание основы этого повреждения. Таким образом, UBIs являются в достаточной мере отличимыми от нейрофибриллярных клубков, тел Леви, включений глиальных клеток (характерных для множественной системной атрофии), кортикальных баллонных клеток и различных поражений, характеризующих другие нейродегенеративные расстройства позднего дебюта, так что их (UBIs) можно рассматривать как определяющие какой-либо конкретный путь дегенеративного процесса. БАС является наиболее знакомым синдромом, который происходит из подобного пути, но предполагается, что другие синдромы представляют собой продолжение спектра клинических проявлений, возникающего из специфической анатомической восприимчивости каждого индивидуального больного.
Остановимся отдельно на анатомо-патологических коррелятах клинических фенотипов.
Прогрессирующая мышечная атрофия. Данный фенотип обозначает очевидную дегенерацию нижних моторных нейронов при отсутствии признаков поражения верхних моторных нейронов. Дифференциальный диагноз вбирает в себя синдром Кеннеди (Х-связанная спинобульбарная мышечная атрофия) и взрослые формы спинальной мышечной атрофии; обе патологии характеризуются генетическими расстройствами. Больные с синдромом Кеннеди имеют увеличенный регион повтора цитозин-аденин-гуанина в гене, кодирующем андрогенный рецептор (кодируемый в Х-хромосоме), и случаи спинальной моторной атрофии имеют все более и более выраженную взаимосвязь с делециями в smn- и naip-генах (кодируемых в хромосоме 5). Исходя из этого, диагностическая путаница может быть преодолена непосредственно при помощи генетических средств. P.G. Ince (2000) выполнил посмертные исследования 2 больных с синдромом Кеннеди и 4 больных с болезнью Werdnig — Hoffman (инфантильная спинальная моторная атрофия). Ни в одном из этих случаев убиквитинированные включения не были выявлены, причем во множественных срезах, исследованных на всем протяжении моторной системы и также в головном мозге. В серии 90 случаев БАС-болезни моторного нейрона (ALS-MND), выполненных P.G. Ince (2000) при аутопсии, были 14 больных, у которых признаки поражения верхних моторных нейронов не были клинически выявлены ни на одной из стадий. Из них в 12 случаев убиквитинированные включения найдены на каком-либо определенном уровне спинного мозга или в группах бульбарных моторных нейронов. Также было 2 исключения: 1 больной с необычной семейной историей и пожилой больной с вялотекущей 10-летней историей прогрессирующей слабости. Представляется вероятным, что оба этих больных не имели истинного сочетания БАС — болезнь двигательного нейрона (ALS-MND).
Существенным аспектом у подобных больных с прогрессирующей мышечной атрофией является степень надежности и достоверности клинических методов обнаружения дисфункции верхних моторных нейронов на поздней стадии заболевания, когда наблюдается тяжелая повсеместная слабость и амиотрофия. Возможность того, что дегенерация кортикоспинального тракта может быть замечена при подобных обстоятельствах, в настоящее время изучается. Целый ряд методов задействуется для выявления дегенерации длинного тракта в тканях аутопсии. В соответствии с опытом автора, ни традиционное миелиновое окрашивание, ни метод пропитывания Marchi не являются такими же чувствительными, как иммуноцитохимическое окрашивание на активную микроглию и макрофаги. Из 12 случаев, упомянутых выше, эти разнообразные методы показали субклиническое вовлечение в патологический процесс кортикоспинального тракта у 7 больных. Данный тип наблюдения подчеркивает трудности, возникающие при классифицировании случаев сочетания ALS-MND (БАС — болезнь моторного нейрона) на основе одних лишь клинических проявлений. Многие, но не все, случаи прогрессирующей мышечной атрофии демонстрируют некоторую степень прогрессирования по направлению к БАС, выявляемую при аутопсии, а молекулярная патология дегенерации моторных нейронов в большинстве случаев прогрессирующей мышечной атрофии неразличима от патологии, характерной для БАС.
Первичный боковой амиотрофический склероз. Первичный боковой амиотрофический склероз представляет собой медленно прогрессирующий спастический парапарез; его с меньшей степенью уверенности можно связать с БАС исходя из клинических и патологических исследований. Дифференциальная диагностика включает исключение у пациента наследственного спастического парапареза, рассеянного склероза, тропического парапареза, ассоциированного с вирусом-1 типа Т-клеточной лейкемии человека, и миелопатии, вызванной вирусом иммунодефицита человека. Патологические сведения редки, и их давно повторно рассмотрел ряд исследователей (Konagaya et al., 1998). Десять случаев первичного бокового склероза были заявлены в литературных источниках с 1977 г. как БАС (Konagaya et al., 1998; Fisher, 1977; Beal, Richardson, 1981; Kuzahara et al., 1985), и, кроме того, исследователями изучался еще 1 дополнительный случай, также отнесенный к БАС (Lowe, неопубликованные наблюдения, исследование головного мозга). Во всех, кроме 3 из этих случаев наблюдалась существенная атрофия префронтальной извилины, а также ассоциированная гибель тела клеток Беца. Большинство из случаев (7 из 11) были зафиксированы до начала применения иммуноцитохимии убиквитина, позволяющей выявлять убиквитинированные включения (UBI) и тельца Буниной, но в 3 случаях наблюдались эозинофильные включения внутри клеток переднего рога или ядра подъязычного черепно-мозгового нерва. Среди других случаев (Shaw et al., 1998) можно упомянуть 2, в которых типичные убиквитиновые включения были идентифицированы в нескольких нижних двигательных нейронах при аутопсии.
В случае, исследованном J. Lowe и P.G. Ince (неопубликованные наблюдения, исследование спинного мозга), не были получены доказательства вовлечения нижних двигательных нейронов ни при иммуноцитохимическом исследовании множественных срезов со многих спинальных уровней, ни при детализированном количественном измерении численности моторных нейронов, которые выполнялись на основе ранее сообщавшихся методов оптимального подсчета. Эти данные согласуются с предположением о том, что случаи первичного бокового склероза представляют собой спектр дегенерации, протекающей по типу БАС, предстающей в виде расстройства верхних моторных нейронов, при котором возможно позднее субклиническое расширение, способное вовлекать и нижние моторные нейроны. Дополнительной существенной особенностью подобных случаев является наличие убиквитинированных включений в церебральной коре, которые наделены распределительным паттерном, типичным для случаев БАС-деменции (Lowe, неопубликованные наблюдения), что позволяет предположить, что случаи первичного бокового склероза также вливаются в спектр БАС-деменции. Однако известны некоторые различия, состоящие в том, что дегенерация черного вещества при первичном боковом склерозе может быть малозаметна и не выражена (Konagaya et al., 1998). Два случая изучения белков теплового шока, исследованных автором, не показали подтверждения наличия ни убиквитиновых включений, ни телец Буниной во всех исследованных участках, и они очевидным образом отличаются от первичного бокового склероза (PLS).
Деменция с включением болезни двигательного нейрона. Синдромы лобно-височной деменции (например, деменция по типу синдрома лобной доли (Mann et al., 1993) и деменция, при которой имеется недостаток в убедительных гистологических характеристиках (Knopmann et al., 1990)), были подразделены на несколько гистопатологических групп (Cooper et al., 1995). В пропорциональном соотношении подобных случаев молекулярная патология церебральных полушарий идентична патологии, которая наблюдается при БАС-деменции. Данное наблюдение привело к возникновению термина «деменция с включением болезни двигательного нейрона», характеризующего такие случаи.
Первоначальные патологические сообщения были основаны на изучении больных, у которых при исследованиях выявлялась только клиника расстройств головного мозга при полном отсутствии клинических проявлений, имевших отношение к спинальной локализации заболевания. Исходя из подобных неполных аутопсий продолжал оставаться спорным и дискуссионным факт того, являлось ли на самом деле подобное расстройство истинной частью спектра, характерного для БАС и его вариантов. В более давнее время анализировались 4 случая деменции с включением болезни двигательного нейрона (Lowe, неопубликованные наблюдения), в которых исследовался спинной мозг. Все эти случаи продемонстрировали наличие некоторых типичных убиквитинированных включений в клетках переднего рога или в ядре подъязычного нерва, несмотря на недостаточное количество клинических признаков амиотрофии. Любопытно, что в этих случаях не были получены доказательства дегенерации кортикоспинального тракта или вовлечения (причастности к патологическому процессу) моторного кортекса. В дальнейших 3 случаях лобно-височной деменции с церебральными включениями по БАС-типу 1 случай показал как умеренное побледнение кортикоспинального тракта, так и убиквитинированные включения в клетках переднего рога (Holton et al., 1999). Подобное сочетание тяжелой степени когнитивной дисфункции наряду с субклинической патологией нижних двигательных нейронов выглядит как еще один стереотипический паттерн избирательной восприимчивости в пределах конкретной группы больных. Случаи деменции с включением болезни двигательного нейрона также демонстрируют расширение патологического распространения убиквитинированных включений вплоть до уровня базальных ганглиев, хотя похожие изменения также были засвидетельствованы и у больного с БАС-деменцией.
В лобно-височном кортексе больных с деменцией с включением болезни двигательного нейрона существуют нитевидные дистрофические нейриты, показывающие иммунореактивность к убиквитину; они негативны к тау- и методам нейрофиламентного окрашивания. Эти поражения схожи по строению с нейритами, описанными в болезни Хантингтона (Cammarata et al., 1993) и деменции с тельцами Леви (Dicson et al., 1991), хотя у них различается паттерн распространения.
Дополнительная поддержка, помогающая выявить прямую взаимосвязь между БАС и деменцией с включением болезни двигательного нейрона, приходит из примеров семейной формы заболевания. Так, в родственной группе, описанной исследователем Джексоном и коллегами (Jackson et al., 1996), заболевание присутствовало в каждом из 4 поколений семейства и затронуло 9 из 15 членов семейства. Из этих лиц у 6 индивидуумов выявили чистую лобно-височную деменцию (патологически доказанную деменцию с включением болезни двигательного нейрона в единственном случае аутопсии), тогда как у 1 лица был выявлен БАС без признаков деменции, у 1 больного диагностирован БАС с паркинсонизмом, и еще у одного была выявлена БАС-деменция. Это ясно указывает на то, что отдельный единичный аутосомно-доминантный локус может попеременно вызывать у какого-либо семейства как БАС, так и деменцию с включением болезни двигательного нейрона (вместе с перекрывающимися синдромами).
Спектр БАС и взаимосвязь с вариантами БАС. При клиническом фенотипе БАС в огромной мере доминирует дегенерация моторной системы, и в этой связи патологическое участие в процессе заболевания других областей ЦНС обычно не обнаруживается, если только это не является предметом научных исследований. Подобные экстрамоторные свойства могут зачастую возникать поздно в ходе заболевания, что создает дополнительные трудности их обнаружения. Данное представление поддерживается обнаружением широко распространенной дегенерации ЦНС в тех участках головного мозга, которые классически не подвергаются воздействиям БАС у японских больных на искусственной вентиляции легких, выживающих в течение длительного времени, выходя за рамки той стадии, при которой респираторная дисфункция обычно приводит к летальному исходу (Mizurati et al., 1992). Среди когорты больных с «классическим» БАС существует спектр мультисистемной вовлеченности, которая, вероятно, сливается с изменениями, ассоциированными с БАС-деменцией. С патологической точки зрения БАС-деменция «вливается» и встраивается в «чистую» деменцию с включением болезни двигательного нейрона и в первичный боковой склероз. В схожей мере многие больные с клиническим фенотипом прогрессирующей мышечной атрофии обнаруживают неявную дегенерацию верхних моторных нейронов, выявляемую при аутопсии. Исходя из этой точки зрения, БАС предстает в виде частого клинико-патологического синдрома, который распознается наилучшим образом в границах непрерывного спектра (варьирующегося от чистой прогрессирующей мышечной атрофии, с одной стороны, до первичного бокового склероза и деменции с включением болезни двигательного нейрона, с другой) и возникает из общего патогенетического каскада. Исследования молекулярной патологии позволяют предположить, что наилучшими маркерами данного дегенеративного процесса являются убиквитинированные включения и тельца Буниной.
Семейный БАС. Различные нарушения моторной системы могут быть наследственными, включая:
a) аутосомно-доминантную наследственность (семейный БАС, наследственный спастический парапарез);
b) аутосомно-рецессивную наследственность (спинальная моторная атрофия, наследственный спастический парапарез); и
c) наследственность, обусловленную Х-хромосомой (синдром Кеннеди).
Как было описано выше, патологические признаки наследственного спастического парапареза, прогрессирующей мышечной атрофии и болезни Кеннеди в достаточной степени отличаются от БАС, чтобы указывать на различный патологический механизм, ответственный за нейродегенерацию. Тем не менее до 10% больных БАС обнаруживают позитивную семейную историю, что, как правило, указывает на аутосомно-доминантную наследственность. В этой связи трудно делать однозначные выводы на основе большей части предшествующих литературных данных по причине неопределенности критерия, который позволил бы точно определять семейные заболевания. Отсутствие четкой семейной истории БАС не является неопровержимым доказательством, говорящим в пользу спорадического заболевания. Единственными группами, среди которых уместно проводить окончательные сравнения, являются группы, известные как имеющие мутации в гене, кодирующем SOD1, и группы, демонстрирующие нормальный (здоровый) SOD1-генотип. В этой связи литературные данные относительно патологии семейных видов БАС необходимо рассматривать в двух частях. Литературные сведения о патологии случаев семейных БАС до 1994 г. не включают данные, полученные из анализа SOD1-гена, и в связи с этим их трудно соотносить с нынешними наблюдениями за случаями, в которых генные дефекты полностью охарактеризованы. Было бы очень ценно получить ретроспективный генетический анализ многих случаев, о которых сообщалось в более старой литературе. Самые недавние литературные сведения рассматривают патологию в случаях с четко определенными «каузальными» (причинными) генетическими мутациями. Из практических соображений к данной группе в настоящее время относятся только случаи с мутацией SOD1-гена.
Патология случаев семейного БАС с неохарактеризованной генетической основой. Более ранние литературные источники предполагали, что дегенерация задних столбов спинного мозга чаще замечалась в семейных случаях (Hirano et al., 1967). Предположение о том, что спиноцеребеллярное вовлечение или деменция (Hadson, 1981) имеют слишком большую репрезентацию в семейной форме БАС, не находит широкой поддержки в опубликованных исследованиях. Однако в литературных источниках не существует крупных сравнительных исследований экстрамоторной патологии при семейных БАС и спорадических заболеваниях. О семейных случаях БАС также сообщалось, что они наделены нейропатологическими свойствами, которые идентичны со спорадической формой БАС. Было продемонстрировано, что убиквитинированные включения присутствуют как в спорадических, так и в семейных случаях.
БАС, связанный с мутациями в SOD1-гене. P.G. Ince (2000) сообщает о как минимум 19 случаях, в которых удалось охарактеризовать расстройство SOD1-гена и в которых были выполнены нейропатологические исследования. Степень патологической экспертизы варьировалась, и условия для полностью сопоставимых выводов обеспечивались не всегда. Нет данных о каком-либо последовательном патологическом фенотипе. Многие образцы обладают типичным распределением патологии по типу БАС без существенного немоторного вовлечения. Однако также существует значительное количество случаев, в которых заболевание отчетливо носит мультисистемный характер (Ince, 2000; Живолупов и др., 2011). Во многих из таких случаев выявлена бледность задних столбцов, и этот факт чаще распространен в случаях с мутацией SOD1-гена, чем в спорадических случаях заболевания. Убиквитинированные включения в гиппокампе были выявлены в SOD1-гене семейной формы БАС в случаях, которые не были известны как клинически проявляющие признаки деменции (Ince, 2000).
Главная особенность этих сообщений заключается в наглядности и выраженности нейрофиламентных включений гиалинового конгломерата, которые отмечаются в семейных случаях БАС с мутацией SOD1-гена. Этот вид включения особенно связывается с A4V- и I113T-мутациями, но также есть данные, отметившие его и при H48Q-мутации (Shaw et al., 1997; Ince, 2000). Другие заявленные случаи обнаруживали типичные убиквитиновые включения, и в небольшом количестве случаев содержались оба типа тел включения. Принимая во внимание значительную гетерогенность исследуемых клинических и патологических фенотипов как среди различных SOD1-мутаций, так и при единичной мутации, эти молекулярные патологические наблюдения выглядят необычайно согласованно и последовательно по отношению к A4V и I113T. Другим поразительным примером последовательного фенотипа применительно к какой-либо конкретной SOD1-мутации можно назвать факт ограниченной вовлеченности в патологический процесс кортикоспинального тракта в случаях с A4V-мутацией. Это также обнаруживается при аутопсии, хотя и присутствует некоторая вариабельность в степени вовлечения кортикоспинального тракта.
Бесплатный фрагмент закончился.
Купите книгу, чтобы продолжить чтение.