
Введение
Биохимия — это наука о структуре молекул, входящих в состав живых организмов, а также о механизмах их превращений внутри организма или клетки.
Термин «биохимия» эпизодически употреблялся с середины XIX века, в классическом смысле он был предложен и введён в научную среду в 1903 году немецким химиком Карлом Нейбергом.
Биохимия является продолжением органической химии. Собственно многие молекулы (спирты, гетероциклы и другие) являются объектами исследований специалистами в области органической химии.
Биохимия изучает структуру, свойства и функции молекул, входящих в состав живых организмов. Эту часть биохимии изучает раздел, который получил название «Структурная биохимия». Кроме этого существует второй раздел, получивший название «Биохимия метаболизма». Все молекулы, которые входят в состав клеток и организмов превращаются друг в друга в ходе множества химических реакций. Именно поэтому клетка — это сложнейшая химическая система, состоящая из множества молекул, которые постоянно вступают в различные реакции, в результате образуются другие биологические молекулы. Совокупность всех этих реакций получила название метаболизм. Все реакции метаболизма подразделяют на две большие группы: катаболизм и анаболизм.
Реакции катаболизма — это реакции энергетического обмена, то есть химических превращений органических молекул в энергию АТФ. АТФ или аденозинтрифосфат является основным источником энергии для транспортных, механических и других клеточных процессов. Реакции энергетического обмена направлены на синтез молекул АТФ.
Реакции анаболизма — это реакции пластического обмена, направленные на синтез молекул, из которых собираются компоненты клетки, в результате она растет и размножается. Часто одна молекула превращается в другую не результате одной реакции, в результате их последовательности. Эти последовательности реакций называют метаболитическими путями.
Данный учебник называется «Биохимия метаболизма». В нем рассматриваются последовательности реакций основных метаболитических путей пластического и энергетического обменов, а также механизмы регуляции скорости этих метаболитических путей.
Термодинамика биохимических реакции
Клетка состоит из множества различных молекул, часть из них обнаруживается у всех живых организмов, тогда как другая часть уникальна для клетки и/или отдельного организма. Соответственно, необходимо эти вещества синтезировать. В каждой клетке в одну единицу времени происходит множество химических реакций. Совокупность всех реакций клетки получило название — метаболизм. В каком-то приближении клетку можно сравнить с реактором, в которой происходит множество реакций, из чего можно считать клетку химической системой. Для описания таких систем и сформулированы законы термодинамики.
Первый закон термодинамики гласит: внутренняя энергия системы вместе с ее окружением остается постоянной. Это одна из формулировок закона сохранения энергии, согласно которой можно утверждать, что при любых изменениях системы внутренняя энергия не утрачивается и не приобретается. Вместе с тем внутри рассматриваемой системы энергия может переходить от одной ее части к другой или превращаться из одной формы в другую. Например, химическая энергия может переходить в тепло, превращаться в электрическую энергию, энергию излучения или в механическую энергию.
Второй закон термодинамики гласит: энтропия системы при самопроизвольных процессах возрастает.
Энтропия служит мерой неупорядоченности, хаотичности системы и достигает максимума, когда система приходит в истинное равновесие. При постоянных температуре и давлении соотношение между изменением свободной энергии системы (ΔG) и изменением энтропии (ΔS) представляется следующим выражением, которое объединяет оба закона термодинамики:
ΔG= ΔH-Т ΔS.
где ΔG — изменение свободной энергии системы, то есть та часть изменения внутренней энергии системы, которая может превращаться в работу, ΔН — изменение энтальпии (теплоты), Т — абсолютная температура.
В условиях, при которых протекают биохимические реакции, ΔН приблизительно равно ΔЕ-изменению внутренней энергии системы в результате реакции. В этих условиях приведенное выше выражение можно записать в виде:
ΔG= ΔЕ-Т ΔS
Если ΔG отрицательно, то реакция протекает самопроизвольно и сопровождается уменьшением свободной энергии. Такие реакции называют экзергоническими. Если к тому же ΔG велико по абсолютной величине, то реакция идет практически до конца и ее можно рассматривать как необратимую. Если же ΔG положительно, то реакция будет протекать только при поступлении свободной энергии извне; такая реакция называется эндергонической. Если к тому же ΔG велико, то система является устойчивой и реакция в этом случае практически не осуществляется.
При ΔG равном нулю система находится в равновесии. Причем ферменты не влияют на ΔG реакции ΔG=-RTlnKeq. Таким образом сдвинуть равновесие можно либо сообщая системе дополнительную энергию (проще всего нагреванием) либо увеличивая концентрацию реагентов. Реакция может протекать спонтанно только при отрицательном значении изменения свободной энергии (ΔG). ΔG не зависит от пути, по которому идет реакция, и зависит только от природы реагирующих веществ и их активности (которую можно иногда примерно определять по их концентрации).
Изменение свободной энергии реакции в условиях, когда активность реагирующих веществ и образующихся продуктов равна единице, называется изменением стандартной свободной энергии (ΔG0). Измеряют изменение свободной энергии в стандартных условиях, то есть при давлении 1 атмосфера, 2980 Кельвина (или 250 С), а так же концентрации реагирующих веществ одинаковы и равны 1М. В биологических системах данные параметры не соблюдаются, особенно относительно концентраций реагирующих веществ, в природе концентрация реагирующих веществ никогда и не соответствует стандартным, именно поэтому в биохимии используется понятие ΔG0, которое соответствует понятию свободной энергии в физической химии.
Если рассмотреть реакцию А + В ↔ C + D.
ΔG этой реакции дается уравнением
ΔG = ΔG0 + RTln ([C] [D] / [A] [B])
где ΔG°- изменение стандартной свободной энергии, R-газовая постоянная, Т-абсолютная температура, [А], [В], [С] и [D] -молярные концентрации (точнее активности) реагирующих веществ. ΔG0-изменение свободной энергии реакции при стандартных условиях, когда каждое из реагирующих веществ А, В, С и D присутствует в концентрации 1,0 М. Таким образом, ΔG реакции зависит от природы реагирующих веществ.
Можно легко вывести соотношение между стандартной свободной энергией и константой равновесия реакции. В состоянии равновесия ΔG = 0. Уравнение тогда приобретает следующий вид:
0 = ΔG0 + RTln ([C] [D] / [A] [B])
ΔG0 = -RTlnKeq
Ферменты не влияют на константу равновесия, следовательно, ускоряют только самопроизвольно идущие реакции, где ΔG меньше нуля. Но в клетке множество реакции особенно в процессах биосинтеза изменение свободной энергии больше нуля, чтобы эта реакция прошла необходимо сообщать энергию системе, что в химии чаше всего происходит за счет нагрева системы, в биологических системах это невозможно из-за прежде всего денатурацию белка. Поэтому в биологических системах появился обходной путь: система сопряженных реакций. В одной точке пространства (в данном случае каталитическом центре) происходят одновременно две реакции: у одной ΔG положительно, у другой отрицательно. Если суммарное ΔG отрицательно то обе реакции идут спонтанно. Именно так обеспечивается приток энергии в систему. Наиболее распространенной сопряженной реакцией является гидролиз АТФ.
Главный определитель хода реакции — свободная энергия (ΔG)
В клетке есть очень нужная реакция 1, но ΔG1> 0 — реакция 1 не идет, поэтому в каталитическом центре есть сопряженная реакция 2, ΔG2 <0 — реакция 2 идет. В каталитическом центре находятся субстраты обеих реакций.
Пойдет ли реакция 1 в этой точке определяется суммарным изменением свободной энергии в данной системе (каталитическом центре): Σ ΔG = ΔG1 +ΔG2;
если Σ ΔG> 0 — реакция 1 не идет
если Σ ΔG <0 — реакция 1 идет.
Необходимых реакций для клетки, чья сводная энергия больше нуля очень много, но вторая сопряженная реакция, как донор энергии для системы должна быть унифицирована. Следовательно, необходима реакция 2 (сопряженная) — универсальная и обеспечивающая Σ ΔG <0. Так сложилось в процессе эволюции, что универсальной сопряженной реакцией стала реакция гидролиза АТФ.
АТФ — это универсальная энергетическая валюта в биологических системах, представляет собою богатую энергией молекулу, что обусловлено наличием в ней двух ангидридных связей. Электростатическое отталкивание между этими отрицательно заряженными группами уменьшается при гидролизе ATФ. AДФ и Фн, стабилизируются под действием резонанса в большей степени, чем АТФ. Гидролиз АТФ сдвигает равновесие сопряженной реакции примерно в 108 раз.
Кроме того, АТФ достаточно устойчивая молекула и время ее жизни достаточно велико. Таким образом, для обеспечения процессов биосинтеза клетка постоянно нуждается в притоке энергии — АТФ. В ходе синтеза организм переводит более окисленные вещества в менее окисленные, для чего необходимы доноры протонов и электронов: NADH, NADPH и FADH2.
Совокупность реакций окисления различных биомолекул (углеводов, липидов, аминокислот, нуклеотидов) направленных на синтез АТФ и восстановление NAD+, NADP+ и FAD+ получила название — энергетический метаболизм клетки или катаболизм. Тогда как совокупность реакций биосинтеза биологических молекул и сборки из них клеточных компонентов называют пластическим обменом или анаболизмом.
Таким образом общий метаболизм клетки можно разделить на две большие части: катаболизм и анаболизм.
Энергетический обмен клетки подразделяется в зависимости от класса окисляемых соединений на несколько направлений:
окисление углеводов;
окисление липидов;
окисление белков.
Белки выполняют множество других важных функций, поэтому расщепляются реакциях энергетического обмена в небольших количествах или в случае дефицита других молекул источников энергии. Основными молекулами вступающими в реакции катаболизма являются углеводы и липиды (триацилглицериды).
Углеводы являются одним из основных источников энергии для клетки причем выделяют два пути окисления углеводов: бескислородное (или анаэробное) и с участием кислорода (аэробное). К анаэробным путям окисления углеводов относятся гликолиз, пентозофосфатный шунт и разнообразные виды брожения. Гликолиз является не только путем окисления моносахаридов в параллельным синтезом АТФ, но и прелюдией к путям аэробного окисления или клеточному дыханию.
Рассмотрим пути окисления углеводов более подробно.
Гликолиз
Гликолиз — это совокупность реакций превращения глюкозы в пируват. У аэробных организмов гликолиз служит как бы прелюдией к циклу трикарбоновых кислот и цепи переноса электронов, в ходе которых запасается большая часть свободной энергии, содержащейся в глюкозе. Открытие гликолиза последовало непосредственно за экспериментами Бюхнера, а также Гардена и Ионга по сбраживанию сахара дрожжевым соком. Вскоре с изучением спиртового брожения слились исследования другого направления, связанные с изучением мышц. Физиологи заинтересовались процессом, благодаря которому изолированная мышца могла получать энергию для сокращения в отсутствие кислорода. Хилл показал, что энергию обеспечивает превращение гликогена в лактат, а несколько позднее Мейергоф продемонстрировал, что происходящие при этом химические реакции сходны с теми, которые наблюдаются при спиртовом брожении. Установление структуры изучению гликолиза, проведенными Эмбденом во Франкфурте и Парнасом в Польше. Таким путем вскоре была выяснена последовательность реакций гликолиза (путь Эмбдена — Мейергофа — Парнаса). Все ферменты, катализирующие отдельные стадии процесса, к настоящему времени выделены, закристаллизованы и подробно изучены. Все десять реакций гликолиза протекают в гиалоплазме.
Основным моносахаридом поступающим в гликолиз является глюкоза. В животной клетке присутствуют как свободная глюкоза (поступившая через мембрану из внешней среды), так и продукт распада гликогена (животного полисахарида — мономером которого является глюкоза).
Вся последовательность реакций гликолиза может быть разбита на четыре стадии (последовательность реакций гликолиза представлена на рисунке 1):
подготовка к разрыву цепи;
разрыв цепи и установление равновесия между триозофосфатами;
окислительное образование АТФ
превращение 3-фосфоглицерата в пируват.
Первый этап — подготовка к разрыву цепи:
В гликолиз могут поступать различные свободные шестиуглеродные моносахариды (гексозы) глюкоза, фруктоза и другие, а также глюкоза из гликогена. В первой реакции происходит фосфорилирование молекулы глюкозы ее осуществляет фермент гексокиназа во всех органах и тканях или глюкокиназа в печени. Оба фермента относятся к классу трансфераз, и осуществляют перенос фосфатной группы с молекулы АТФ на молекулу глюкозы с образованием глюкозо-6-фосфата, АТФ в данном случае является как донором энергии, так и донором фосфатной группы, за счет затраты АТФ происходит, во-первых, образование активной формы моносахарида, глюкозо-фосфата, во-вторых, обеспечивается необратимость реакций гликолиза. Разница между ферментами заключается в специфичности (глюкокиназа более специфична взаимодействует только с глюкозой, гексокиназа менее специфична фосфорилирует все гексозы); в распределении в организме (глюкокиназа в печени, гексокиназа в остальных тканях организма); в регуляции (смотри ниже). Второй путь входа через глюкозо-1-фосфат. Так входят в гликолиз глюкоза из гликогена и галактоза.
Образовавшийся фруктозо-6-фосфат под действием фосфофруктокиназы превращается 1,6-фруктозобифосфат. Фосфофруктокиназа также относится к классу трансфераз. Донором энергии и фосфатной группы в этой реакции является молекула АТФ. То есть затрачивается еще одна молекула АТФ. Это делает последовательность реакции гликолиза окончательно необратимой, а кроме того образуется симметричная молекула, что важно для второго этапа гликолиза.
Вторая стадия — разрыв цепи и установление равновесия между триозофосфатами
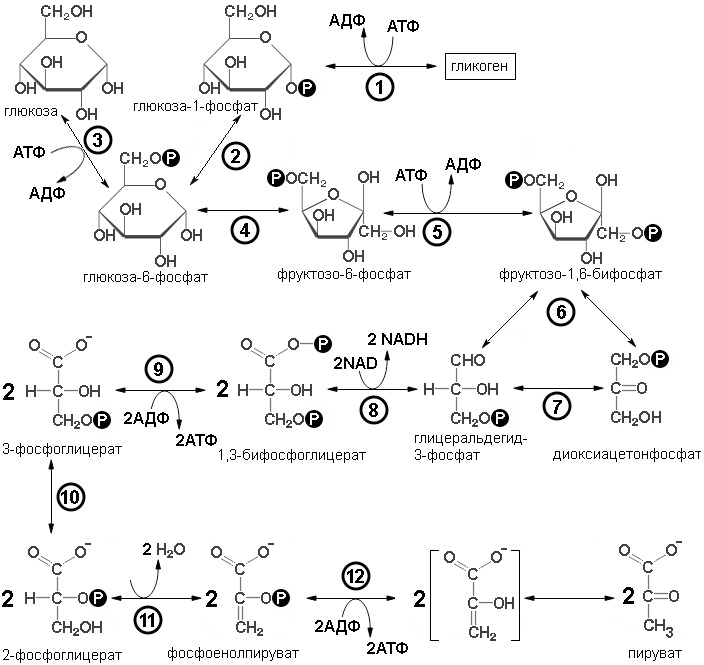
Гликоген — это запасающий полисахарид животных состоящий из α-D-глюкозы. Под действием гликоген фосфорилазы происходит отщепление остатков глюкозы от полимерной цепи гликогена, параллельно происходит присоединение фосфатной группы к молекуле глюкозы с образованием глюкозо-1-фосфата, донором и энергии, и фосфатной группы является молекула АТФ. Через образование глюкозо-1-фосфата в гликолиз поступает галактоза. К галактозе присоединяется УТФ с образованием УДФ-галактозы, которая под действием эпимеразы превращается в УДФ-глюкозу. УДФ-глюкоза распадается на глюкозо-1-фосфат и УМФ, данную реакцию катализирует УДФ-глюкозопирофосфорилаза. Образовавшийся глюкозо-1-фосфат изомеризуется в глюкозо-6-фосфат фосфоглюкоизомеразы. Таким образом образуется активная форма глюкозы глюкозо-6-фосфат. Затем происходит изомеризация глюкозо-6-фосфата в фруктозо-6-фосфат под действием изомеразы. Фруктозо-6-фосфат может образовываться и из свободной фруктозы под действием гексокиназы.
После образования 1,6-фруктозобифосфата начинается вторая стадия гликолиза. Расщепление фруктозодифосфата катализируется альдолазой, относящейся к классу лиаз; в результате образуются глицеральдегид-3-фосфат и диоксиацетонфосфат. Между этими двумя триозофосфатами в результате действия изомеразы устанавливается равновесие. Таким образом, обмен обеих половинок гексозы может пойти по пути превращения в пируват через глицеральдегид-3-фосфат. В то же время для диоксиацетонфосфата существует и другой путь, связанный с восстановлением в глицерофосфат — предшественник липидов и в промежуточный продукт в некоторых типах брожения. Но в случае гликолиза дигидроксиацетонфосфат под действием триозофосфатизомеразы легко преобразуется в глицероальдегид-3-фосфат, и между этими продуктами устанавливается равновесие, но глицеральдегид-3-фосфат, постоянно изымается в следующие реакции гликолиза, поэтому равновесие сдвигается в сторону изомеризации дигидроксиацетонфосфата в глицероальдегид-3-фосфат, и это происходит практически количественно, поэтому считается, что весь дигидроксиацетонфосфат преобразуется в глицероальдегид-3-фосфат, и принято удваивать продукты всех последующих реакций. Глицероальдегид-3-фосфат поступает в третью стадию.
Третья стадия — окислительное образование АТФ
Глицеральдегид-3-фосфат окисляется под действием глицеральдегид-3-фосфат оксидоредуктазы фосфорилирующей акцептором электронов является NAD+, восстанавливающийся до NADH. В результате образуется короткоживущий промежуточный продукт 1,3-бифосфоглицерат, вторая фосфатная группа поступает из раствора. 1,3-бифосфоглицерат является нестабильным соединением, причем ΔG гидролиза связи между карбоксильной и фосфатной групппировками в 1 положении меньше нуля, а по модулю больше энергии гидролиза фосфоангидридной связи АТФ. Такие соединения называют макроэргическими. 1,3-бифосфоглицерат распадается под действием 1,3-бифосфоглицераткиназы и отщепляемая фосфатная группа переносится на молекулу АДФ и в результата образуются 3-фосфоглицерат и АТФ. Такой тип синтеза АТФ называют субстратным фосфорилированием. То есть фосфатная группа переносится с макроэргического соединения (энергия гидролиза фосфатной группы по модулю больше энергии гидролиза АТФ, поэтому выделившейся энергии достаточно для фосфорилирования АДФ и образования АТФ). Образовавшийся 3-фосфоглицерат поступает в 4-й этап гликолиза.
Четвертая стадия — превращение 3-фосфоглицерата в пируват
3-фосфоглицерат, изомеризуется в 2-фосфоглицерат под действием 3—2 фосфоглицерат изомеразы. 2-фосфоглицерат дегидратируется енолазой (2-фосфоглицератгидролиазой), происходит отщепление молекулы воды образуется фосфоенолпируват (ФЕП), который является «макроэргическим» соединением, фосфорильная группа которого может быть легко перенесена на AДФ (под действием фермента пируваткиназы); остающийся при этом енол пировиноградной кислоты самопроизвольно превращается в значительно более устойчивый пируват. Поскольку на каждую молекулу глюкозы образуются две молекулы фосфоенолпирувата, этот процесс восполняет затрату двух молекул АТФ, происходящую на начальных стадиях образования фруктозо-1,6-дифосфата из глюкозы.
Суммарная реакция гликолиза:
Глюкоза +2АТФ +2NAD +4АДФ = 2Пируват +2АДФ +4АТФ +2NADH
Суммарный энергетический выход всего 2 АТФ.
Энергетическая выгода гликолиза не велика, и используется организмами либо имеющими доступ большим количествам субстратов (моносахаридов), например паразитические организмы, либо из-за условий среды (анаэробные организмы).
Гликолиз — это один из древнейших метаболитических путей, по некоторым данным считается первые живые организмы получали энергию путем гликолиза. Поэтому механизмы регуляции гликолиза очень хорошо отрегулированы и направлены на обеспечение клетки энергией с одной стороны, но при этом на сохранение ресурсов клетки с другой. Механизмы регуляции гликолиза изучены достаточно хорошо.
Регуляция гликолиза
Регуляция гликолиза происходит на трех этапах:
Вход глюкозы в гликолиз (это естественно, так как если процесс не нужен, то его проще не запускать вообще, а не обрывать на половине).
Фосфофофруктокиназная реакция (реакция необратима, кроме того в ней затрачивается АТФ).
Пируваткиназная реакция (реакция также необратима, а кроме того важным является процесс утилизации образующегося пирувата).
Теперь необходимо рассмотреть эти этапы более подробно.
Вход глюкозы в гликолиз
Как было рассмотрено выше, глюкоза входит в гликолиз из свободной глюкозы или из гликогена. Свободная глюкоза фосфорилируется гексокиназой, активность этого фермента регулируется: происходит ингибирование продуктом реакции глюкозо-6-фосфатом. Поэтому накопление глюкозо-6-фосфата резко снижает скорость гексокиназной реакции, в результате нет затрат АТФ, так как глюкозо-6-фосфат во всех тканях кроме печени направляется на реакции окисления. Тогда как в печени накопление глюкозо-6-фосфата не происходит, так как излишки запасаются в виде гликогена, поэтому в печени работает другой фермент — глюкокиназа, не ингибируемый продуктом реакции.
При входе глюкозы из гликогена первая реакция гликогенфосфорилазная, регуляция гликогенфосфорилазы происходит двумя путями. Первый вариант посттрансляционная модификация.
В скелетных мышцах этот фермент присутствует в двух формах — в каталитически активной фосфорилированной форме (фосфорилаза а) и в значительно менее активной дефосфорилированной форме (фосфорилаза b) (схема перехода изорм представлена на рисунке 2). Фосфорилаза а была получена в кристаллическом виде (мол. масса 190 кDa). Ее молекулы состоят из двух идентичных субъединиц, каждая из которых содержит существенный для каталитической активности остаток серина в фосфорилированной форме. Скорость превращения структурных единиц гликогена в глюкозо-1-фосфат регулируется в мышцах соотношением активной фосфорилазы а и менее активной фосфорилазы b. Взаимопревращения двух этих форм гликогенфосфорилазы происходят под действием специфичных ферментов, катализирующих процесс ковалентной модификации фосфорилазы. Фосфорилаза а превращается в менее активную фосфорилазу b под действием фермента, называемого фосфатазой фосфорилазы а; этот фермент, катализируя гидролитический разрыв связей, удаляет из молекулы фосфорилазы а фосфатные группы, необходимые для каталитической активности.

Фосфорилаза b вновь превращается в активную фосфорилазу а под действием фермента, называемого киназой фосфорилазы b; он катализирует реакцию, в ходе которой АТФ фосфорилирует остатки серина в активном центре молекулы фосфорилазы b, что и приводит к образованию фосфорилазы а. Таким образом, благодаря действию двух ферментов, фосфатазы фосфорилазы а и киназы фосфорилазы b, соотношение активной фосфорилазы а и сравнительно мало активной фосфорилазы b в клетке может изменяться. В мышцах действует второй механизм регуляции гликогенфосфорилазной активности. Фосфорилаза b, сравнительно мало активная форма, может становиться более активной в результате нековалентного связывания с аллостерическим модулятором этого фермента, которым является AMФ; концентрация же AMФ в мышцах возрастает по мере распада АТФ в сократительных системах. Активации фосфорилазы b под действием AMФ препятствует АТФ, выступающий в роли отрицательного модулятора. Таким образом, активность фосфорилазы b определяется соотношением AMФ и ATФ. В отличие от фосфорилазы b фосфорилаза а не активируется AMФ; поэтому фосфорилазу а называют иногда AMФ-независимой формой, а фосфорилазу b AMФ-зависимой.
Таким образом, есть два механизма регуляции, которым подчиняется гликогенфосфорилаза скелетной мышцы: 1) ковалентная модификация посредством фосфорилирования или дефосфорилирования остатков серина в активном центре фермента и 2) аллостерическая регуляция фосфорилазы b путем нековалентного связывания с AMФ или АТФ. В покоящейся мышце почти вся фосфорилаза находится в неактивной, или b -форме, поскольку в такой мышце концентрация АТФ. В печени гликогенфосфорилаза также присутствует в а- и b-форме; в принципе ферменты печени функционируют подобно мышечным, от которых они, впрочем, несколько отличаются по своей структуре и регуляторным свойствам. Расщепление гликогена в печени имеет иное назначение, нежели в мышцах; этот процесс служит источником свободной глюкозы крови. Под действием фосфорилазы печени образуется глюкозо-1-фосфат, который затем превращается в глюкозо-6-фосфат, являющийся уже непосредственным предшественником свободной глюкозы. Реакция, в ходе которой образуется D-глюкоза крови, катализируется ферментом глюкозо-6-фосфатазой.
Второй этап регуляции гликолиза — регуляция реакции образования фруктозо-1,6-дифосфата, катализируемой фосфофруктокиназой. Фосфофруктокиназа (ФФК) — это сложный аллостерически регулируемый фермент, управляемый многими аллостерическими положительными и отрицательными модуляторами. В скелетных мышцах активность фосфофруктокиназы определяется концентрациями субстратов этого фермента (АТФ и фруктозо-6-фосфата) и его продуктов (AДФ и фруктозо-1,6-дифосфата); все эти соединения играют роль аллостерических регуляторов. Очень важны также в качестве регуляторов AMФ, цитрат, ионы Mg2+, фосфат и некоторые другие метаболиты, присутствующие в мышечной ткани. Однако, хотя регуляции ФФК зависит от сложного взаимодействия ряда факторов, главными отрицательными модуляторами этого фермента являются АТФ и цитрат, а самыми активными положительными модуляторами AMФ и фруктозо-1,6-дифосфат. Всякий раз, когда при очень активном мышечном сокращении концентрация АТФ падает, а энергии требуется больше, фосфофруктокиназная активность усиливается, даже если концентрация фруктозо-6-фосфата очень низка. Если, однако, уровень АТФ в клетке уже высок по сравнению с уровнем AДФ и AMФ, то кажущееся сродство фосфофруктокиназы к фруктозо-6-фосфату сильно. В этом случае фосфофруктокиназа будет катализировать реакцию лишь при сравнительно высокой концентрации фруктозо-6-фосфата Цитрат, один из промежуточных продуктов цикла лимонной кислоты, усиливает ингибирование фосфофруктокиназы высокими концентрациями АТФ. В то же время повышение концентрации AMФ, образующегося в результате аденилаткиназной реакции в сокращающейся мышце, служит очень мощным стимулирующим модулятором и противодействует ингибирующему влиянию АТФ на фосфофруктокиназную реакцию.
В результате всех этих сложных аллостерических взаимодействий скорость реакции, катализируемой фосфофруктокиназой, возрастает иногда в сотни раз при переходе скелетной мышцы из состояния покоя к состоянию максимальной активности.
Третьим регулируемым этапом гликолиза является пируваткиназная реакция. Пируваткиназа также принадлежит к числу аллостерических ферментов. Этот фермент встречается, по меньшей мере, в трех изоформах, которые отличаются друг от друга по распределению в тканях и по реакции на различные модуляторы. При высоких концентрациях АТФ кажущееся сродство пируваткиназы к фосфоенолпирувату сравнительно невелико и соответственно невелика скорость пируваткиназной реакции при обычных концентрациях фосфоенолпирувата. Пируваткиназу ингибируют также ацетил-СоА и высокомолекулярные жирные кислоты — соединения, играющие важную роль в качестве топлива для цикла лимонной кислоты. Таким образом, когда в клетке уже велика концентрация АТФ или когда в ней уже достаточно топлива для процесса дыхания, обеспечивающего клетку энергией, гликолиз ингибируется за счет либо фосфофруктокиназы, либо пируваткиназы (в зависимости от условий). В то же время при низких концентрациях АТФ кажущееся сродство пируваткиназы к фосфоенолпирувату возрастает, и это позволяет ферменту переносить фосфатные группы от фосфоенолпирувата на AДФ даже при относительно низкой концентрации фосфоенолпирувата. Некоторые аминокислоты также действуют как модуляторы пируваткиназной активности, главным образом в печени. Во всех клетках гликолиз регулируется с очень высокой эффективностью, напоминающей действие компьютера, а потому изменения концентрации различных метаболитов могут влиять на его общую скорость.
Гликолиз — процесс анаэробного окисления моносахаридов, в результате которого происходит синтез АТФ. У аэробных организмов АТФ синтезируется еще и в процессах клеточного дыхания, в которых утилизируются другие продукты гликолиза (пируват и NADH), поэтому направление реакций гликолиза сдвинуто в сторону образования продуктов метаболитического пути.
У анаэробных организмов гликолиз один из основных путей энергетического обмена углеводов, но утилизации остальных продуктов не происходит, накопление этих продуктов может привести к остановке гликолиза, а следовательно и остановке энергетического обмена (АТФ не образуется), поэтому необходимо утилизировать пируват и NADH. Для этого появилась «надстройка» — брожение, для окисления NADH и сдвига реакций в сторону образования пирувата.
Брожение
У аэробных организмов пируват и NADH, образовавшиеся в ходе гликолиза, утилизируются в ходе клеточного дыхания. У анаэробных организмов этого не происходит, поэтому необходима надстройка. Этой надстройкой является процесс брожения.
Брожение — это тип анаэробного окисления пирувата.
В настощее время описано множество путей брожения. Все они выявлены у прокариотических организмов. Но два типа бружения встречаются не только у прокариот, но и у эукариот. У эукариотических организмов обнаружены гомоферментативное молочнокислое и спиртовое брожение.
Гомоферментативное молочнокислое брожение
Гомоферментативное молочнокислое брожение получило свое название (схема представлена на рисунке 3), из-за того, что происходит одна реакция осуществляемая ферментом лактат дегидрогеназой молекула пирувата восстанавливается до лактата и донором протонов и электронов является NADH.
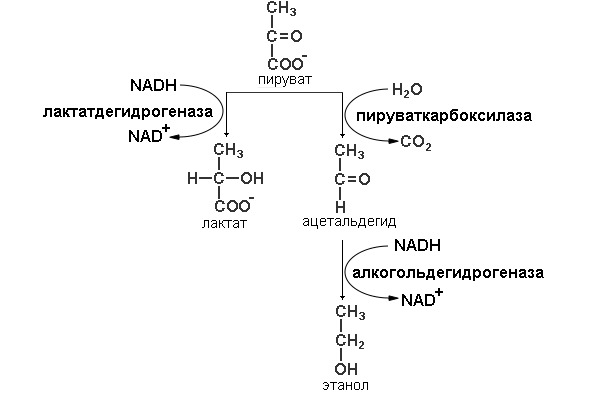
Данный тип брожения характерен для многих бактерий. Многие возбудители заболеваний, например, стафилококки и стрептококки, осуществляют этот тип брожения. Но данный брожения встречается и у полезных бактерий. Молочнокислые бактерии используются при изготовлении молочнокислых продуктов, сыров, сырокопченых колбас, при квашении капусты, засолке огурцов, силосовании при заготовке кормов. Также молочнокислое брожение встречается и у эукариот. Паразиты крови (простейшие, паразитирующие в крови млекопитающих (возбудители малярии, сонной болезни), также используют данный тип утилизации пирувата.
Лактатдегидрогеназа присутствует и в тканях млекопитающих. Хотя в обычных условиях наши мышцы получают вполне достаточные количества кислорода, чтобы произошло окисление пирувата и образование АТФ аэробным путем, бывают обстоятельства, когда поступление кислорода оказывается недостаточным. Например, при крайнем напряжении сил, когда уже весь запас кислорода израсходован, мышечные клетки образуют лактат путем брожения. Более того, в белых мышцах рыб или домашней птицы аэробный метаболизм относительно невелик, и основным конечным продуктом оказывается L-лактат. В организме человека есть такие ткани, которые слабо снабжаются кровью, например, хрусталик и роговица глаза. В клетках этих тканей окислительный метаболизм выражен слабо, а энергия в основном образуется при сбраживании глюкозы в лактат. Часть лактата, образующегося в мышцах и других тканях, поступает в кровь и переносится в печень, где он снова окисляется в пируват. Меньшая часть пирувата затем окисляется в цикле трикарбоновых кислот, но большая его часть снова превращается в глюкозу. Последняя может опять поступать в кровь и возвращаться в мышцы. Весь этот процесс называется циклом Кори.
Спиртовое брожение
У дрожжей и у других микроорганизмов, сбраживающих глюкозу не до лактата, а до этанола и СО2, путь ферментативного расщепления глюкозы совпадает с описанным выше для анаэробного гликолиза на всем протяжении, за исключением этапа, катализируемого лактатдегидрогеназой (схема представлена на рисунке 3). В дрожжевых клетках, которые не содержат фермента, аналогичного лактатдегидрогеназе мышечной ткани, этот этап заменен двумя другими реакциями. В первой из них продукт расщепления глюкозы пируват теряет свою карбоксильную группу под действием пируватдекарбоксилазы. Эта реакция представляет собой простое декарбоксилирование; реального окисления пирувата при этом не происходит. Для проявления каталитической активности пируватдекарбоксилазе требуется Mg2+. С молекулой этого фермента прочно связан кофермент тиаминпирофосфат. На последнем этапе спиртового брожения ацетальдегид восстанавливается до этанола за счет NADH, образовавшегося при окислении глицеральдегид-3-фосфата; эта реакция катализируется алкогольдегидрогеназой.
Таким образом, конечными продуктами спиртового брожения являются этанол и СО2, а не лактат. Пируватдекарбоксилаза содержится в клетках пивных дрожжей и других микроорганизмов, осуществляющих спиртовое брожение. В животных тканях этот фермент отсутствует. Лишены пируватдекарбоксилазы также организмы, осуществляющие молочнокислое брожение, например молочнокислые бактерии. Биохимия спиртового брожения лишь недавно изучена настолько хорошо, чтобы можно было представить этот процесс в виде ряда последовательных ферментативных реакций.
Что же касается виноделия и пивоварения, то это весьма древние искусства, освоенные людьми за сотни лет до того, как родилась сама наука химия. Более того, сами старинные рецепты приготовления пива и вина сыграли в свое время важную роль, послужив ключом к некоторым фундаментальным открытиям на заре развития биологии и биохимии.
Так, в 1856 г. Луи Пастер впервые убедительно показал, что сбраживание сахара в спирт вызывается микроорганизмами, а не какими-то магическими влияниями. Французские виноделы пригласили Пастера для того, чтобы он помог им выяснить, почему в иные годы вино не удается и превращается в уксус. Пастер в своих экспериментах, ставших классическими, показал, что в стерильных растворах глюкозы брожения не происходит, тогда как в растворах, соприкасающихся с нефильтрованным воздухом, брожение идет, и причина этого заключается в том, что в раствор попадают из воздуха споры дрожжей и других микроорганизмов.
Из налета на гроздьях свежесрезанного винограда Пастер выделил культуры дрожжей и доказал, что именно дрожжи ответственны за брожение, происходящее в соке, отжатом из раздавленного винограда. Он выяснил также, что превращение спирта в уксусную кислоту вызывается другими видами микроорганизмов — уксуснокислыми бактериями; эти аэробные организмы окисляют этанол с образованием уксусной кислоты.
Моносахариды вообще и глюкоза в частности используются не только для генерирования энергии в ходе гликолиза, но и в других метаболических путях, причем как в катаболических так и анаболических. К таким цепям реакций относятся: пентозофосфатный путь и окисление глюкозы до аскорбиновой кислоты.
Пентозофосфатный путь
Пентозофосфатный путь является альтернативным путем окисления глюкозы. Он включает несколько этапов, в результате функционирования которых из трех молекул глюкоза-6-фосфата образуются три молекулы СО2 и три молекулы пентоз. Последние используются для регенерации двух молекул глюкозо-6-фосфата и одной молекулы глицеральдегид-3-фосфата. Поскольку из двух молекул глицеральдегид-3-фосфата можно регенерировать молекулу глюкоза-б-фосфата, глюкоза может быть полностью окислена при превращении по пентозофосфатному пути.
У растений часть реакций пентозофосфатного пути участвует также в образовании гексоз из СО2 при фотосинтезе. Пентозофосфатный путь называют иногда пентозным шунтом, гексозомонофосфатным путем или фосфоглюконатным окислительным путем. Открытие Отто Варбургом (Otto Warburg) в 1931 г. глюкозо-6-фосфат-дегидрогеназы, первого фермента этого пути, сделало возможной его полную расшифровку, которую осуществили Фриц Липман, Фрэнк Дикенс, Бернард Хорекер и Эфроим Рэкер.
Пентозофосфатный цикл не приводит к синтезу АТР, он выполняет две главные функции: 1) образование NADPH для восстановительных синтезов, таких, как синтез жирных кислот и стероидов; 2) обеспечение рибозой синтеза нуклеотидов и нуклеиновых кислот. Ферменты пентозофосфатного пути локализованы во внемитохондриальном пространстве клеткив цитозоле. Как и в процессе гликолиза, окисление осуществляется путем дегидрогенирования, однако акцептором водорода в этом случае служит не NAD, а NADP.
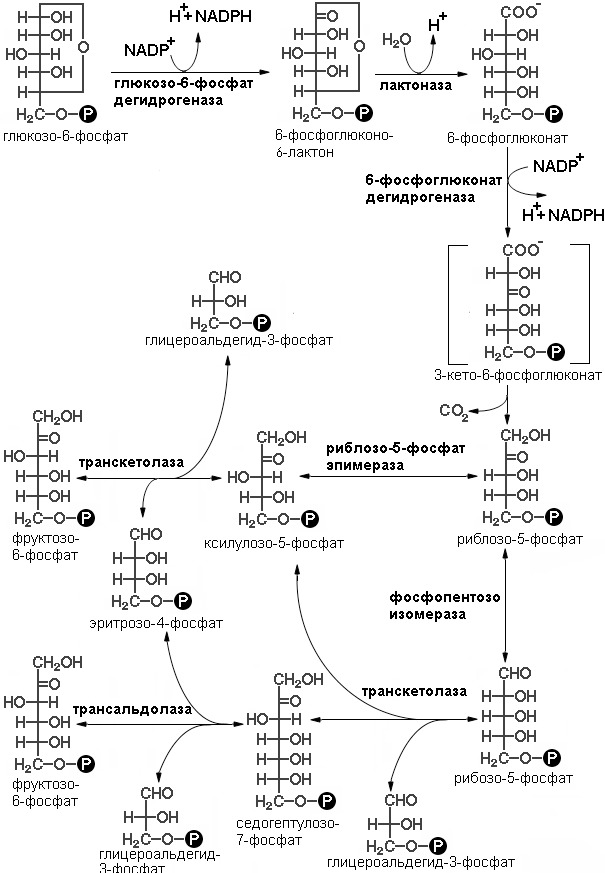
Последовательность реакций пути можно разделить на две фазы: окислительную и неокислительную (схема реакций представлена на рисунке 4). В реакциях первой фазы глюкоза-6-фосфат дегидрогенируется и декарбоксилируется с образованием рибулозо-5-фосфата. В ходе второй фазы рибулозо-5-фосфат превращается снова в глюкозо-6-фосфат в результате серии реакций, в которых главную роль играют два фермента: транскетолаза и трансальдолаза
Окислительная фаза пентозофосфатного пути начинается с дегидрирования глюкозо-6-фосфата при С-1, реакции, катализируемой глюкозо-6-фосфат-дегидрогеназой.
Фермент высокоспецифичен в отношении NADP+; Км для NAD+ примерно в тысячу раз выше, чем для NADP+. Продуктом реакции является 6-фосфоглюконо-δ-лактон, внутримолекулярный эфир, с эфирной связью между С-1-карбоксильной группой и гидроксилом при С-5. Следующий этап — гидролиз 6-фосфоглюконо- δ -лактона специфической лактоназой, дающий 6-фосфо-глюконат. Этот шестиуглеродный сахар подвергается затем окислительному декарбоксилированию 6-фосфоглюконат — дегидрогеназой с образованием рибулозо-5-фосфата. Акцептором электронов вновь служит NADP+. (смотри рисунок). Конечным этапом синтеза рибозо-5-фосфата является изомеризация рибулозо-5-фосфата фосфопентозо-изомеразой. Эта реакция подобна гликолитическим реакциям:
Глюкозо-6-фосфат ↔ Фруктозо-6-фосфат
Дигидроксиацетонфосфат ↔ Глицеральдегид-3-фосфат.
Все три кетозо-альдозные изомеризации идут через образование ендиольного промежуточного продукта.
Окислительная фаза пентозофосфатного пути иногда считается основной и неокислительная рассматривается как связка с гликолизом. Это связано с тем фактом, что эти фазы могут идти как независимо друг от друга, так вместе. В ходе неокислительной фазы пентозофосфатного пути происходит регенерация рибозо-5-фосфата в глюкозо-6-фосфат. Основную роль в этом процессе играют два фермента: транскетолаза и трансалъдолаза. Эти же ферменты создают обратимую связь между пентозофосфатным путем и гликолизом, катализируя следующие три реакции:
Транскетолаза переносит двухуглеродную группу, включающую 1-й и 2-й атомы углерода кетозы, на альдегидный углерод альдозного сахара. Происходит, следовательно, превращение кетосахара в альдозу, содержащую на два атома углерода меньше, и одновременное превращение альдосахара в кетозу, содержащую на два атома углерода больше. Коферментом реакции является тиаминидифосфат (в его состав входит тиамин — витамин группы В), для ее протекания необходимы ионы Mg2+. Переносимая двухуглеродная группа является, вероятно, гликоальдегидом, связанным с тиаминдифосфатом, т. е. «активным гликольальдегидом». Транскетолаза катализирует перенос двухуглеродной группы с ксилулозо-5-фосфата на рибозо-5-фосфат с образованием семиуглеродной кетозы седогептулозо-7-фосфата и альдозы глицеральдегид-3-фосфата. Эти два продукта далее вступают в следующую реакцию, называемую трансальдолазной. Трансальдолаза осуществляет перенос трехуглеродного фрагмента, «активного дигидроксиацетона» (атомы углерода 1 — 3), кетозы седогептулозо-7-фосфата на альдозу глицеральдегид-3-фосфат; в результате образуются кетоза фруктоза-6-фосфат и четырехуглеродная альдоза эритрозо-4-фосфат. Следующая реакция снова катализируется транскетолазой. В этой реакции ксилулозо-5-фосфат служит донором «активного гликоальдегида». Роль акцептора выполняет образовавшийся ранее эритрозо-4-фосфат. Продуктами этой реакции являются фруктоза-6-фосфат и глицеральдегид-3-фосфат.
Итак, избыток рибозо-5-фосфата, образованный в пентозофосфатном пути, может количественно превращаться в промежуточные продукты гликолиза.
Значение метаболического пути для различных тканей можно оценить по его активности. Пентозофосфатный путь активно протекает в печени, жировой ткани, коре надпочечников, щитовидной железе, эритроцитах, семенниках и в молочных железах в период лактации; он неактивен в нелактирующей молочной железе и малоактивен в скелетных мышцах. Все ткани, в которых активность данного пути высока, используют в реакциях восстановительного синтеза NADPH, например в реакциях синтеза жирных кислот, стероидов, аминокислот (с участием глутаматдегидрогеназы) или восстановленного глутатиона в эритроцитах. Вероятно, в условиях активного липогенеза или при наличии любой системы, утилизирующей NADPH, возрастает активная деградация глюкозы по пентозофосфатному пути в связи с увеличением отношения NADP+/NADPH. В условиях, которые возникают после приема пищи, может индуцироваться синтез глюкоза-6-фосфатдегидрогеназы и 6-фосфоглюконатдегидрогеназы.
Регуляция скорости функционирования пентозофосфатного пути
Первая реакция окислительной ветви пентозофосфатного пути, дегидрирование глюкозо-6-фосфата, по существу необратима. Действительно, при физиологических условиях эта реакция лимитирует скорость процесса и выполняет функцию «контрольного пункта». Наиболее важным регуляторным фактором является концентрация NADP+, акцептора электронов при окислении глюкозо-6-фосфата в 6-фосфоглюконо-лактон. Кроме того, NADPH конкурирует с NADP+ за связывание с ферментом, и АТР конкурирует с глюкозо-6-фосфатом. Отношение концентрации NADP+ к концентрации NADPH в цитозоле печени крыс, содержащихся на полноценном рационе, составляет примерно 0,014, что на несколько порядков ниже отношения [NAD+] / [NADH], которое при этих же условиях равно 700. Выраженное действие концентрации NADP+ на скорость превращений по окислительной ветви пентозофосфатного пути подтверждает, что генерирование NADPH тесно сопряжено с его использованием в восстановительных биосинтезах. Вопрос о регуляции неокислительной ветви пентозофосфатного пути до сих пор остается открытым.
Регуляция направления пентозофосфатного шунта
Судьба глюкозо-6-фосфата зависит от потребности в NADPH, рибозо-5-фосфате и АТФ.
В данном случае возможно четыре различные ситуации (схема регуляторных механизмов представлена на рисунке 5).
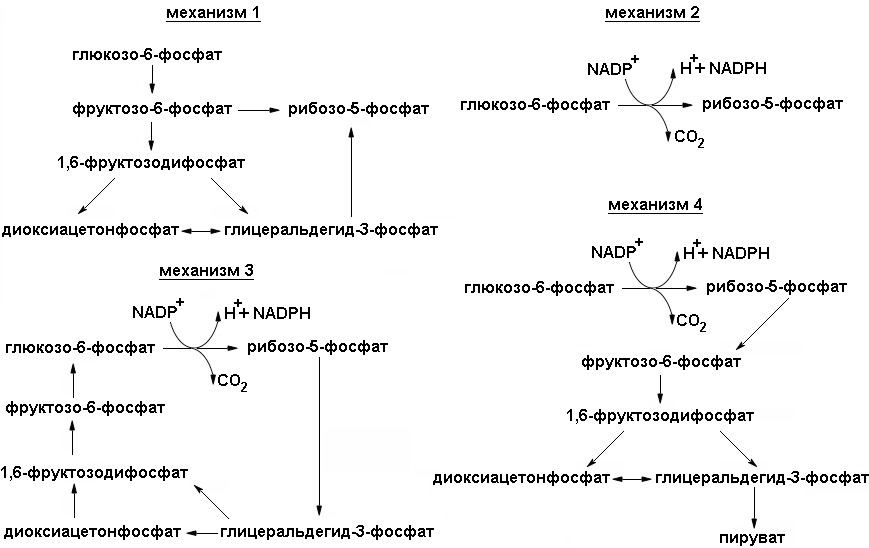
1. Потребность в рибозо-5-фосфате значительно превышает потребность в NADPH. Большая часть глюкозо-6-фос-фата превращается во фруктозо-6-фостфат и глицеральдегид-3-фосфат по гликолитическому пути. Затем две молекулы фрукто-зо-6-фосфата и одна молекула глицераль-дегид-3-фосфата превращаются под действием трансальдолазы и транскетолазы в три молекулы рибозо-5-фосфата путем обращения реакции, описанной ранее. Стехиометрия этого превращения следующая:
5Глюкозо-6-фосфат +5АТР → 6Рибозо-5-фосфат +5ADP + Н +.
Из чего можно заключить, что в данном случае идет только неокислительная фаза.
2. Потребность в NADPH и рибозо-5-фосфате сбалансирована. При таких условиях преобладающей реакцией является образование двух молекул NADPH и одной молекулы рибозо-5-фосфата из одной молекулы глюкозо-6-фосфата по окислительной ветви пентозофосфатного пути. Стехиометрия этого превращения описывается уравнением
Глюкозо-6-фосфат +2NADP+ + Н2О → Рибозо-5-фосфат +2NADPH +2Н+ + СО2.
Из чего можно заключить, что в данном случае идет только окислительная фаза.
3. Потребность в NADPH значительно превышает потребность в рибозо-5-фосфате; глюкозо-6-фосфат полностью окисляется в С02. В этой ситуации активно протекают три группы реакций. Во-первых, по окислительной ветви пентозофосфатного пути образуются два NADPH и один рибозо-5-фосфат. Далее рибозо-5-фосфат превращается во фруктозо-6-фосфат и гли-церальдегид-3-фосфат под действием транс-кетолазы и трансальдолазы. Наконец, происходит ресинтез глюкозо-6-фосфата из фруктозо-6-фосфата и глицеральдегид-3-фосфата по пути глюконеогенеза (рассматривается ниже в этой главе). Стехиометрия указанных реакций описывается следующими уравнениями:
6Глюкозо-6-фосфат +12NADP+ +6Н2О → 6Рибозо-5-фосфат +12NADPH +12Н+ +6СО2,
6Рибозо-5-фосфат → 4Фруктозо-6-фосфат +2Глицеральдегид-3-фосфат,
4Фруктозо-6-фосфат +2Глицеральдегид-3-фосфат + Н2О → 5Глюкозо-6-фосфат + Фн
Суммируя эти реакции, получается
Глюкозо-6-фосфат +12NADP+ +7Н2О → 6СО2 +12NADPH +12Н+ + Фн
Таким образом, эквивалент глюкозо-6-фосфата может быть полностью окислен до С02 с одновременным генерированием NADPH. Смысл указанных реакций состоит в том, что рибозо-5-фосфат, образовавшийся по пентозофосфатному пути, вновь превращается в глюкозо-6-фосфат под действием транскетолазы, трансальдолазы и некоторых ферментов глюконеогенеза. То есть происходят обе фазы пентозофосфатного пути.
4. Потребность в NADPH значительно превышает потребность в рибозо-5-фосфате: глюкозо-6-фосфат превращается в пируват. Возможен и другой путь; рибозо-5-фосфат образовавшийся по окислительной ветви пентозофосфатного пути, превращается в пируват. Фруктозо-6-фосфат и глицеральдегид-3-фосфат, происходящие из рибозо-5-фосфата, вступают на гликолитический путь обмена, а не подвергаются обратному превращению в глюко-зо-6-фосфат. Согласно изложенному механизму, происходит одновременное генерирование АТР и NADPH и пять из шести атомов углерода глюкозо-6-фосфата появляются в пирувате:
3Глюкозо-6-фосфат +6NADP+ +5NAD+ +5Фн +8AДФ → 5Пируват +3СО2 +6NADPH +5NADH +8АТФ +2Н2О +8Н +.
Образовавшийся в этих реакциях пируват может окисляться с образованием дополнительного количества АТФ или может быть использован в качестве строительного блока в различных биосинтетических процессах.
Превращение глюкозы в глюкуроновую и аскорбиновую кислоты
По другому вторичному пути катаболизма глюкозы в животных тканях образуются два специализированных продукта: D-глюкуронат, важная роль которого связана с обезвреживанием и выведением из организма чужеродных органических веществ, и L-аскорбиновая кислота (витамин С) (схема реакций образования аскорбиновой кислоты представлена на рисунке 6).
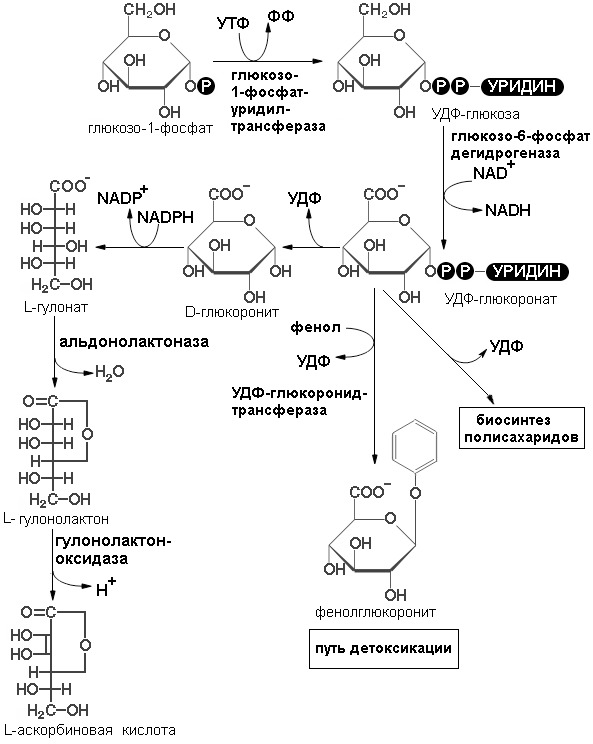
В этом случае D-глюкозо-1-фосфат сначала взаимодействует с УТФ и превращается в УДФ-глюкозу. Затем глюкозная часть молекулы УДФ-глюкозы подвергается ферментативному дегидрированию с образованием УДФ-D-глюкуроната. Эта реакция представляет собой еще один пример использования УДФ-производных в качестве промежуточных продуктов при ферментативных превращениях сахаров. УДФ-D-глюкуронат способствует обезвреживанию некоторых чужеродных веществ или лекарственных препаратов (например, фенола) и таким образом усиливает их выведение через почки. Кроме того, УДФ-глюкуронат служит предшественником D-глюкуронатных остатков в структурных полисахаридов, и промежуточным продуктом в процессе превращения D- глюкозы в L-аскорбиновую кислоту. Он восстанавливается за счет NADPH до шестиуглеродной сахарной кислоты L-гулоната, которая затем превращается в соответствующий лактон. L-гулонолактон дегидрируется до L-аскорбиновой кислоты, или витамина С, при участии флавопротеина гулополактон-оксидазы. Именно этим путем синтезируется L-аскорбат в растениях и у тех животных, которые способны обеспечивать себя этим витамином.
В организме человека, морской свинки, обезьян, некоторых видов птиц и индийской плодоядной летучей мыши витамин С не синтезируется; эти виды должны получать его в готовом виде, с пищей. Человек, морская свинка и разные виды обезьян не синтезируют витамин С потому, что у них отсутствует фермент гулонолактон — оксидаза. Можно думать, что некогда все организмы располагали набором ферментов, необходимых для синтеза аскорбата, но затем какие-то виды утратили эту способность к синтезу вследствие мутации, которая, однако, не оказалась для них летальной, поскольку обычную пищу данного вида составляли богатые витамином С растения.
Доля глюкозы, отвлекаемой на этот вторичный путь, очень невелика, по сравнению с большим ее количеством, расщепляемым в процессе гликолиза и через цикл лимонной кислоты. Однако продукты таких вторичных путей жизненно необходимы организму.
Клеточное дыхание
Как говорилось выше, у аэробных организмов пируват, образовавшийся в ходе гликолиза, поступает в процесс клеточного дыхания, который дает значительно больше молекул АТФ, чем гликолиз или другие пути анаэробного окисления углеводов.
Клеточное дыхание включает три стадии:
окислительное образование ацетил-СоА из пирувата, жирных кислот и аминокислот, расщепление ацетильных остатков в цикле лимонной кислоты, в результате которого образуются СО2 и атомы водорода, перенос электронов на молекулярный кислород, сопряженный с окислительным фосфорилированием AДФ до АТФ.
При окислении углеводов гликолиз завершается образованием пирувата, который поступает в пируват дегидрогеназный комплекс, катализирующий образование ацетил-коА, то есть в первый этап клеточного дыхания, где субстратом являются углеводы.
Пируватдегидрогеназная реакция
В аэробных условиях конечный продукт гликолиза пируват подвергается сначала дегидрированию и декарбоксилированию с образованием ацетил-Со А и СО2. Катализирует этот процесс пируватдегидрогеназный комплекс, располагающийся во внутренней мембране митохондрии и состоящий из трех последовательно действующих ферментов, важным коферментом которого является тиамин пирофосфат (ТПФ), производное витамина В1. Недостаток витамина B1, или тиамина, обуславливает заболевание, известное под названием бери-бери.
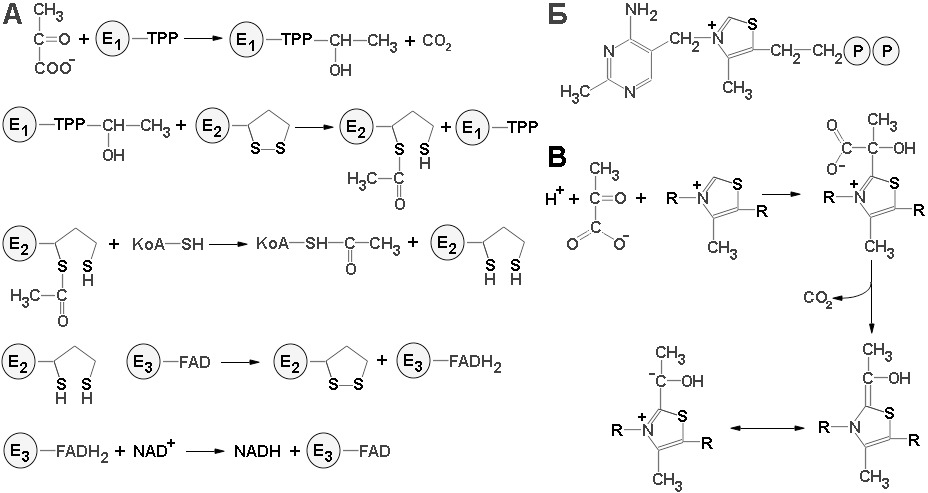
Теперь нам ясно, что в организме животных, лишенных тиамина, оказывается невозможным нормальное окисление пирувата. Особенно сильно влияет такое нарушение на мозг, который обычно получает всю необходимую энергию путем аэробного окисления глюкозы и для которого поэтому окисление пирувата жизненно необходимый процесс. Превращение пирувата в ацетил-СоА происходит в четыре стадии (схема реакций с участием пируватдегидрогеназного комплекса представлен на рисунке 7).
На первой стадии пируват соединяется с ТПФ и затем подвергается декарбоксилированию. Реакция катализируется пируват-дегидрогеназным компонентом мультиферментного комплекса. Решающее значение для данного процесса имеет следующая особенность ТПФ, у простетической группы пируватдегидрогеназного компонента: очень кислый характер атома углерода, находящегося между атомами азота и серы тиазолового кольца (смотри рис. 7, Б). Этот атом углерода ионизируется, образуя карбанион, который легко присоединяется к карбонильной группе пирувата. Положительно заряженный азот в кольце ТПФ принимает на себя электроны, стабилизируя формирование отрицательного заряда, необходимого для декарбоксилирования.
Затем протонирование приводит к образованию гидроксиэтиламинпирофосфата. На второй стадии гидроксиэтильная группа, связанная с ТПФ, окисляется с образованием ацетильной группы и одновременно переносится на липоамид. Окислителем в этой реакции служит дисульфидная группа липоамида, которая превращается в сульфгидрильную группу. Реакция катализируется дигидролипоил трансацетилазным компонентом комплекса и приводит к образованию ацетиллипоамида. На третьей стадии ацетильная группа переносится с ацетиллипоамида на СоА, образуя ацетил-СоА, Процесс также катализируется дигилролипоилтрансацетилазой.
При переходе ацетильной группы на СоА сохраняется богатая энергией тиоэфирная связь. На четвертой, завершающей стадии происходит регенерирование окисленной формы липоамида. Реакция катализируется дигидролипоил-дегидрогеназным компонентам комплекса, Окислителем в ней служит NAD+, а роль простетической группы фермента выполняет FAD+.
Пируватдегидрогеназный комплекс настолько крупный, что по размеру может быть сравнен с рибосомой или другой млекулярной «машиной». Молекулярная масса данного комплекса составляет 4600 кDa и размер 300 А. В состав комплекса входит 48 полипептидных цепей, ядро комплекса образуют трансацетилазные цепи, пируват и липоил дегидрогеназные комплексы присоединяются к ядру с внешней стороны. Структурное объединение трех видов ферментов делает возможным координированный катализ при осуществлении сложной реакции. Все промежуточные продукты реакции окислительного декарбоксилирования пирувата прочно связываются с комплексом. Тесная близость между ферментами увеличивает суммарную скорость процесса и сводит к минимуму побочные реакции. Активированные промежуточные продукты переносятся от одного активного центра к другому липоамидной простетической группой трансацетилазы. Присоединение липоильной группы к ε-аминогруппе лизинового остатка трансацетилазы создает гибкий рычаг для реакционноспособного кольца. Этот молекулярный рычаг в 14Å способствует взаимодействию липоильной части трансацетилазной субъединицы с тиаминпирофосфатным компонентом соседней пируват-дегидрогеназной субъединицы и с флавиновым компонентом соседней липоилдегидрогеназы. Кроме того, липоильные компоненты мультиферментного комплекса могут реагировать друг с другом, образуя сеть взаимодействующих реакционноспособных групп.
Таким образом, суммарная реакция пируватдегидрогеназного комплекса может быть сформулирована следующим образом:
Пируват + NAD+ →AC-coA + CO2+NADH
Скорость реакции пируватдегидрогеназной реакции регулируется.
Регуляция пируватдегидрогеназного комплекса
Реакция образования ацетил-СоА, катализируемая пируватдегидрогеназным комплексом, регулируется в животных тканях при помощи ковалентной модификации этого комплекса. Когда концентрация АТФ в митохондриях относительно велика и когда ацетил-СоА, а также промежуточные продукты цикла Кребса имеются в достаточном количестве, обеспечивающем удовлетворение энергетических нужд клетки, дальнейшее образование ацетил-СоА приостанавливается. В этих условиях, которые служат сигналом для такой приостановки, АТФ является положительным модулятором, активирующим вспомогательный фермент — киназу пируватдегидрогеназы. Этот фермент использует АТФ для фосфорилирования остатка серина в активном центре молекулы пируватдегидрогеназы, в результате чего образуется неактивная форма фермента — фосфопируватдегидрогеназа.
Если, однако, потребность в АТФ возрастает и уровень АТФ соответственно снижается, то неактивная, фосфорилированная, форма пируватдегидрогеназы может быть вновь активирована. Это происходит в результате гидролитического отщепления от молекулы пируватдегидрогеназы ингибирующей фосфатной группы. Катализирует эту реакцию другой фермент — фосфатаза фосфопируватдегидрогеназы. Стимулирующее действие на этот фермент оказывает повышение концентрации ионов Са2+, играющих роль важного метаболического посредника; концентрация ионов Са2+ увеличивается всякий раз, когда возникает потребность в АТФ.
Киназа пируватдегидрогеназы и фосфатаза фосфопируватдегидрогеназы присутствуют в пируватдегидрогеназном комплексе. Этот комплекс, следовательно, представляет собой очень сложную, независимую и саморегулирующуюся систему. Пируватдегидрогеназный комплекс регулируется также путем аллостерической модуляции. Сильное ингибирующее действие оказывают на него (помимо АТФ) ацетил-СоА и NADH, которые являются продуктами пируватдегидрогеназной реакции и в то же время играют роль аллостерических ингибиторов этой системы. Аллостерическое ингибирование окисления пирувата резко усиливается в присутствии высокомолекулярных жирных кислот; ниже будет показано, что жирные кислоты тоже служат источником ацетил-СоА. Таким образом, каталитическая активность пируватдегидрогеназного комплекса выключается в тех случаях, когда в клетках имеется достаточно топлива в виде жирных кислот и ацетил-СоА или когда в них повышаются концентрация АТФ и отношение NADH/NAD+.
Цикл трикарбоновых кислот
Ацетил-СоА поступает в цикл трикарбоновых кислот, который является вторым этапом клеточного дыхания. Впервые предположение о существовании такого цикла для окисления пирувата в животных тканях было высказано в 1937 г. Гансом Кребсом. Эта идея родилась у него, когда он исследовал влияние анионов различных органических кислот на скорость поглощения кислорода суспензиями измельченных грудных мышц голубя, в которых происходило окисление пирувата. Грудные мышцы отличаются чрезвычайно высокой интенсивностью дыхания, что делает их особенно удобным объектом для изучения окислительной активности. Незадолго до описываемых работ Кребса Альберт Сент-Дьёрдьи в Венгрии обнаружил, что некоторые четырехуглеродные дикарбоновые органические кислоты, присутствующие в животных тканях (янтарная, фумаровая, яблочная и щавелевоуксусная), способны усиливать поглощение кислорода мышечной тканью. Кребс подтвердил это наблюдение и показал, что перечисленные органические кислоты стимулируют также окисление пирувата. Кроме того, он нашел, что окисление пирувата мышечной тканью стимулируется шестиуглеродными трикарбоновыми кислотами — лимонной, цис-аконитовой и изолимонной, а также пятиуглеродной α-кетоглутаровой кислотой. Испытаны были и некоторые другие встречающиеся в природе органические кислоты, но ни одна из них не обнаружила подобной активности. Обращал на себя внимание сам характер стимулирующего действия активных кислот: даже малого количества любой из них было достаточно для того, чтобы вызвать окисление во много раз большего количества пирувата.
Цикл трикарбоновых кислот выполняет несколько функций.
Во-первых, цикл трикарбоновых кислот представляет собою конечный общий путь для окисления топливных молекул. Большинство топливных молекул вступают в цикл в виде ацетил-СоА.
Во-вторых, образующиеся в реакциях цикла трикарбоновых кислот активные восстановительные эквиваленты, такие как NADH и FADH2, затем поступают в третий этап — окислительное фосфорилирование, в результате которого образуется основная масса молекул АТФ. Именно поэтому цикл трикарбоновых кислот является одним из основных путей энергетического обмена.
В-третьих, цикл трикарбоновых кислот служит также источником строительных блоков для процессов биосинтеза.
Цикл трикарбоновых кислот осуществляется в митохондрии, где располагаются ферменты, катализирующие реакции этого цикла. Большая часть ферментов располагается в матриксе митохондрии. Исключение — α-кетоглутаратдегидрогеназа и сукцинатдегидрогеназа, располагающиеся во внутренней мембране митохондрии.
Несмотря на то, что реакции замкнуты в цикл, их можно подразделить на два этапа: окисление ацетил коА до СО2 и регенерация оксалоацетата (последовательность реакций цикла трикарбоновых кислот представлена на рисунке 8).
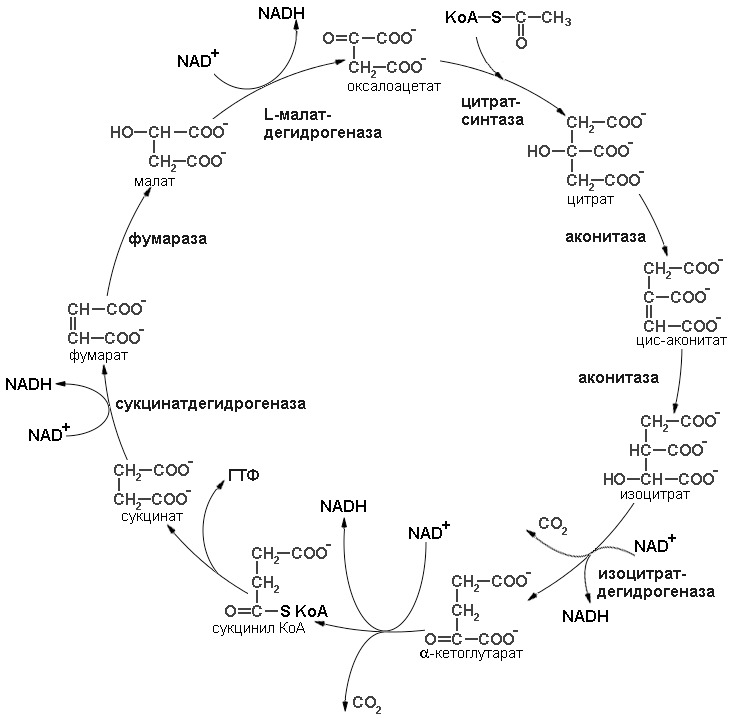
Первая стадия окисления ацетил-СоА начинается с начальной реакции цикла. Начальная реакция — конденсация ацетил-СоА и оксалоацетата, приводящая к образованию цитрата, катализируется конденсирующим ферментом, цитратсинтазой, при этом происходит образование связи углерод-углерод между метильным углеродом ацетил-СоА и карбонильным углеродом оксалоацетата. За реакцией конденсации, приводящей к образованию цитрил-СоА, следует гидролиз тиоэфирной связи, сопровождающийся потерей большого количества свободной энергии в форме теплоты; это определяет протекание реакции слева направо до ее завершения. Превращение цитрата в изоцитрат катализируется аконитазой (аконитатгидратазой), содержащей ион железа в Fe2+-состоянии. Ион железа является кофактором, обеспечивающим правильную организацию каталитического центра. Аконитаза относится классу лиаз, а не изомераз, как могло бы показаться на первый взгляд. Эта реакция осуществляется в две стадии: сначала происходит дегидратация с образованием цис-аконитата (часть его остается в комплексе с ферментом), а затем — гидратация и образование изоцитрата. Реакция ингибируется фторацетатом, который сначала превращается во фторацетил-СоА; последний конденсируется с оксалоацетатом, образуя фторцитрат. Непосредственным ингибитором аконитазы является фторцитрат; при ингибировании накапливается цитрат.
Среди простых соединений самым «смертельным» ядом является фторацетат натрия. LD50 (доза, летальная для 50% получивших ее животных) для крыс составляет всего лишь 0,2 мг/кг — почти в 10 раз меньше летальной дозы нервного яда диизопропилфторфосфата. Широко употребляемый (хотя и отзывы о его действии противоречивы) как яд для грызунов «1080», фторацетат был также обнаружен в листьях некоторых ядовитых растений, встречающихся в Африке, Австралии и Южной Америке. Примечательно, что дифторацетат НСР2—СОО- вообще нетоксичен. Биохимическими исследованиями установлено, что сам фторацетат не оказывает на клетки токсического действия. Токсичность проявляется лишь после метаболического превращения фторацетата в фтороцитрат, являющийся высокоспецифичным ингибитором аконитазы. Этот факт весьма примечателен, поскольку изомерный фторцитрат, образующийся в реакции фтороксалоацетата с ацетил-СоА, оказывает на тот же фермент лишь слабое ингибирующее действие, хотя как раз у этого изомера атом фтора находится на том участке, который атакуется аконитазой. Атом фтора имеет небольшой ван-дер-ваальсов радиус (0,135 нм), сравнимый с ван-дер-ваальсовым радиусом водорода (0,12 нм); на это обстоятельство часто ссылаются, когда говорят о способности фторсодержащих соединений «обманывать» ферменты. Однако более вероятно, что высокая электроотрицательность фтора и его способность к участию в водородных связях делают его в метаболическом отношении более сравнимым с — ОН-группой. В случае с фторцитратом предполагают, что ингибирующий изомер связывается с аконитазой «неправильным способом», при котором атом фтора оказывается координационно-связанным с атомом железа в активном центре аконитазы.
Эксперименты с использованием промежуточных соединений, меченных изотопом С14, показывают, что аконитаза взаимодействует с цитратом асимметрично: она всегда действует на ту часть молекулы цитрата, которая образовалась из оксалоацетата. Это сначала было трудно объяснить, так как лимонная кислота является внешне симметричным соединением. Однако положение в пространстве двух групп — СН-СООН лимонной кислоты относительно групп — ОН и — СООН неидентично. Об асимметричном действии аконитазы свидетельствует «судьбах меченого ацетил-СоА (т. е. положение атомов С14) в интермедиатах цикла лимонной кислоты. Возможно, что цис-аконитат не является обязательным интермедиатом между цитратом и изоцитратом и образуется на боковой ветви основного пути.
Образовавшийся изоцитрат окисляется под действием изоцитратдегидрогеназы до оксалосукцината или α-кетоглутарата. Окислению подвергается гидроксильная группа изоцитрата, образующаяся в результате кето-группа дестабилизирует карбоксильную группу во втором положении, это приводит к декарбоксилированию. Карбоксильная группа отщепляется в виде молекулы углекислого газа. Это первая молекула СО2, образующаяся при окислении ацетил-СоА. Важным компонентом реакции декарбоксилирования являются ионы Мn2+ (или Mg2+). Акцептором протонов и электронов является молекула NAD+, в результате образуется NADH. Описаны три различных формы изоцитратдегидрогеназы. Одна из них, NAD+-зависимая, найдена только в митохондриях. Две другие формы фермента являются NADP+-зависимыми, причем одна из них также находится в митохондриях, а другая в цитозоле. Окисление изоцитрата, связанное с работой дыхательной цепи, осуществляется почти исключительно NAD+-зависимым ферментом.
Образовавшийся в результате окисления изоцитрата α-кетоглутарат подвергается окислительному декарбоксилированию, сходному с окислительным декарбоксилированием пирувата. Окислительное декарбоксилирование α-кетоглутарата катализируется α-кетоглутарат-дегидрогеназным комплексом, сходным по структуре с пируват-дегидрогеназным комплексом. В состав α-кетоглутарат-дегидрогеназного комплекса входят три вида ферментов: α-кетоглутарат-дегидрогеназный, транссукцинилазный и дигидролипоил-дегидрогеназный компоненты. Ядро комплекса составляет транссукцинилаза (аналогично трансацетилазе). α-Кетоглутарат — дегидрогеназный компонент и транссукцинилаза отличаются от соответствующих ферментов пируват-дегидрогеназного комплекса. В то же время дигидролипоил-дегидрогеназные части обоих комплексов идентичны. Видно, что пируват- и α-кетоглутарат-дегидрогеназные комплексы представляют собою гомологичные ассоциации ферментов. Структурные и механистические особенности, обеспечивающие координированный катализ на входе в цикл трикарбоновых кислот, вновь используются позднее в процессе функционирования этого цикла. Равновесие реакции настолько сильно сдвинуто в сторону образования сукцинил-СоА, что ее можно считать физиологически однонаправленной. Как и при окислении пирувата, реакция ингибируется арсенатом, что приводит к накоплению субстрата (α-кетоглутарата). Реакция окисления α-кетоглутарата с образованием сукцинил-СоА является реакцией окислительного декарбоксилирования с образованием параллельно молекул СО2 и NADH. Это последняя реакция первого этапа цикла.
Все последующие реакции относятся ко второму этапу цикла — регенерации оксалоацетата.
Продолжением цикла является превращение сукцинил-СоА в сукцинат, катализируемое сукцинаттиокиназой (сукцинил-СоА-синтетазой). Одним из субстратов реакций является ГДФ (или ИДФ), из которого в присутствии неорганического фосфата образуется ГТФ (ИТФ). Это — единственная стадия цикла лимонной кислоты, в ходе которой генерируется высокоэнергетическая фосфатная связь на субстратном уровне; при окислительном декарбоксилировании α-кетоглутарата потенциальное количество свободной энергии достаточно для образования NADH и высокоэнергетической фосфатной связи. В реакции, катализируемой фосфокиназой, АТФ может образовываться как из ГТФ, так и из ИТФ.
Сукцинаттиокиназа — фермент, располагающийся в печени. В альтернативной реакции протекающей во всех других тканях кроме печени и катализируемой сукцинилСоА-ацетоацетат-СоА-трансферазой (тиофоразой), сукцинил-СоА превращается в сукцинат сопряженно с превращением ацетоацетата в ацетоацетил-СоА. В данной реакции нуклеозидтрифосфаты не образуются, но эта реакция используется для активации ацето-ацетата, что важно для обмена липидов, который будет подробно рассмотрен ниже.
Далее сукцинат дегидрогенируется с образованием фумарата. Дегидрогенирование катализируется сукцинатдегидрогеназой, связанной с внутренней поверхностью внутренней митохондриальной мембраны. Это единственная дегидрогеназная реакция цикла лимонной кислоты, в ходе которой осуществляется прямой перенос водорода с субстрата на флавопротеин без участия NAD+. Фермент содержит FAD+ и железо-серный (Fe-S) белок. В рзультате окисления сукцината до фумарата образуется FADH2. Добавление малоната или оксалоацетата ингибирует сукцинатдегидрогеназу, что приводит к накоплению сукцината.
Фумараза (фумаратгидратаза) катализирует присоединение воды к фумарату с образованием малата. Фумараза специфична к L-изомеру малата, она катализирует присоединение компонентов молекулы воды по двойной связи фумарата в транc-конфигурации. Образовавшийся малат окисляется с образованием оксалоацетата, в результате происходит замыкание цикла трикарбоновых кислот.
Реакцию окисления малата осуществляет малатдегидрогеназа. Малатдегидрогеназа катализирует превращение малата в оксалоацетат, реакция идет с участием NAD+. Хотя равновесие этой реакции сильно сдвинуто в направлении малата, реально она протекает в направлении оксалоацетата, поскольку он вместе с NADH постоянно потребляется в других реакциях.
Ферменты цикла лимонной кислоты, за исключением α-кетоглутарат и сукцинатдегидрогеназы, обнаруживаются и вне митохондрий. Однако некоторые из этих ферментов (например, малатдегидрогеназа) отличаются от соответствующих митохондриальных ферментов.
Таким образом, два атома углерода поступают в цикл в виде ацетил-СоА и два атома углерода покидают цикл в виде СО2 при последовательных реакциях декарбоксилирования.
Суммарная реакция цикла трикарбоновых кислот:
Ac-СoA+3NAD++FAD++ГДФ →2CO2+3NADH+FADH2+ГТФ
Суммарная реакция полного окисления глюкозы до CO2:
Глюкоза +2АТФ +10NAD+ +2FAD+ +4АДФ+2ГДФ→ 6CO2+2АДФ +4АТФ +10NADH+2FADH2+2ГТФ
Регуляция скорости цикла трикарбоновых кислот
Регуляция скорости цикла трикарбоновых кислот осуществляется на уровне регуляции скорости нескольких реакций цикла. В большинстве случаев скорость функционирования метаболических циклов определяется их начальными этапами.
Полагают, что так же обстоит дело и в случае цикла лимонной кислоты. Общая скорость его функционирования во многих тканях определяется первой реакцией: синтезом цитрата. Разумеется, скорость цитратсинтазной реакции регулируется концентрацией ее субстратов, в частности концентрацией ацетил-СоА, а она в свою очередь зависит от активности пируватдегидрогеназного комплекса. Регулируется эта реакция также концентрацией второго субстрата — оксалоацетата; возможно даже, что этот фактор играет главную роль, поскольку концентрация оксалоацетата в митохондриях очень низка и зависит от метаболических условий. На активность цитратсинтазы влияет также концентрация сукцинил-СоА, одного из более поздних промежуточных продуктов цикла. Как только концентрация сукцинил-СоА превышает нормальный стационарный уровень, цитратсинтаза сразу же ингибируется, поскольку сукцинил-СоА понижает ее сродство к ацетил-СоА, то есть сукцинил-СоА является отрицательным аллостерическим регулятором (ингибитором) цитратсинтазы, уменьшающим ее активность. Жирные кислоты, служащие предшественниками ацетил-СоА тоже ингибируют цитратсинтазу, являясь отрицательными аллостерическими эффекторами. В некоторых клетках роль аллостерических ингибиторов цитратсинтазы играют цитрат и NADH.
У большей части клеток окисление изоцитрата до α-кетоглутарата и CO2, которое может происходить под действием двух разных изоцитратдегидрогеназ, регулируется, по-видимому, путем аллостерической стимуляции NAD-зависимого фермента, вызываемой AДФ. В то же время NADH и NADPH действуют как отрицательные аллостерические модуляторы изоцитратдегидрогеназной активности.
Ингибитором активности α-кетоглутаратдегидрогеназного комплекса служит продукт реакции сукцинил-СоА. Таким образом, в цикле лимонной кислоты регулируются, по меньшей мере, три стадии, и только в своих деталях эта регуляция у разных типов клеток несколько различается.
Скорость гликолиза в нормальных условиях согласована со скоростью функционирования цикла лимонной кислоты: в клетке до пирувата расщепляется ровно столько глюкозы, сколько необходимо для того, чтобы обеспечить цикл лимонной кислоты «топливом», т. е. ацетильными группами ацетил-СоА. Ни пируват, ни лактат, ни ацетил-СоА обычно не накапливаются в аэробных клетках в больших количествах; их концентрации поддерживаются на некоем постоянном уровне, соответствующем динамическому равновесию. Согласованность между скоростью гликолиза и скоростью функционирования цикла лимонной кислоты объясняется не только тем, что первый процесс ингибируется высокими концентрациями АТФ и NADH, т. е. компонентами, общими для гликолитической и дыхательной стадий окисления глюкозы; определенную роль в этой согласованности играет также и концентрация цитрата. Продукт первой стадии цикла лимонной кислоты — цитрат является аллостерическим ингибитором фосфофруктокиназы, катализирующей в процессе гликолиза реакцию фосфорилирования фруктозо-6-фосфата.
Механизмы анаплеротических реакций
Цикл лимонной кислоты — это еще и один из амфиболических путей. Он используется не только для окислительного катаболизма, т. е. для расщепления углеводов, жирных кислот и аминокислот, но может служить также первой стадией многих биосинтетических путей, для которых он является источником предшественников. Под воздействием ряда важных вспомогательных ферментов некоторые промежуточные продукты цикла лимонной кислоты, главным образом α-кетоглутарат, сукцинат и оксалоацетат, могут удаляться из цикла и использоваться в качестве предшественников аминокислот. Скорость функционирования цикла лимонной кислоты при этом, казалось бы, должна снижаться, поскольку такой отток промежуточных продуктов из цикла должен понижать их концентрацию в клетке. В действительности же этого не происходит, так как убыль промежуточных продуктов цикла восполняется благодаря действию другого набора ферментов. При нормальных условиях реакции, отвлекающие промежуточные продукты из цикла, и реакции, восполняющие их убыль, находятся в состоянии динамического равновесия, так что концентрация этих продуктов в митохондриях остается более или менее постоянной.
Специальные ферментативные реакции, обеспечивающие пополнение пула промежуточных продуктов цикла лимонной кислоты, носят название анаплеротических («пополняющих») реакций. Наиболее важная реакция такого рода в животных тканях это ферментативное карбоксилирование пирувата за счет СО2 с образованием оксалоацетата катализирует эту обратимую реакцию фермент пируваткарбоксилаза. Если для цикла лимонной кислоты не хватает оксалоацетата или какого-нибудь другого промежуточного продукта цикла, то карбоксилирование пирувата стимулируется и запас оксалоацетата растет. Для ферментативного присоединения карбоксильной группы к молекуле пирувата требуется энергия. Источником ее служит сопряженное с данной реакцией расщепление АТФ до AДФ и фосфата. Поскольку суммарная реакция сопровождается лишь незначительным изменением стандартной свободной энергии, мы можем заключить, что свободная энергия, необходимая для присоединения карбоксильной группы к пирувату, примерно равна свободной энергии, выделяющейся при гидролизе АТФ. Пируваткарбоксилаза очень сложный фермент. Его молекулярная масса равна приблизительно 650000 Da. Молекула фермента содержит четыре кофермента. Каждый из них состоит из одной молекулы витамина биотина, ковалентно связанного (пептидной связью) с аминогруппой особого остатка лизина, находящегося в активном центре. Свободный СО2, предшественник новой карбоксильной группы оксалоацетата, сначала активируется путем присоединения к одному из атомов азота в молекуле биотина. Эта активация, связанная с расходованием АТФ, составляет первую стадию реакции, катализируемой пируваткарбоксилазой. На второй стадии, протекающей также в активном центре фермента, новая карбоксильная группа, ковалентно связанная с простетической группой фермента, переносится на пируват с образованием оксалоацетата. Пируваткарбоксилаза принадлежит к регуляторным ферментам. В отсутствие ацетил-СоА, который служит для нее положительным модулятором, скорость катализируемой ею прямой реакции, приводящей к образованию оксалоацетата, очень невелика. Избыток же ацетил-СоА. поставляющего «топливо» для цикла лимонной кислоты, стимулирует пируваткарбоксилазную реакцию; в результате этого образуется больше оксалоацетата и цикл использует больше ацетил-СоА в цитратсинтазной реакции. Пируваткарбоксилазная реакция — главная анаплеротическая реакция в печени и почках. В миокарде и в мышцах протекают другие анаплеротические реакции. Одна из таких реакций катализируется фосфоенолпируваткарбоксикиназой. В этой реакции происходит расщепление фосфоенолпирувата — сверхвысокоэнергетического фосфорилированного соединения, образующегося в процессе гликолиза. Высвобождаемая энергия используется для карбоксилирования с образованием оксалоацетата, а ее остаток запасается в форме ГТФ.
Глиоксилатный цикл — одна из модификаций цикла лимонной кислоты
У растений и некоторых микроорганизмов, например у Е. coli, ацетильные группы часто служат не только высокоэнергетическим «топливом», но и источником метаболитов, из которых строятся углеродные скелеты углеводов. В таких клетках действуют два варианта цикла лимонной кислоты: 1) обычная последовательность реакций, в ходе которой происходит окисление ацетил-СоА до СО2 свойственная большинству тканей, и 2) особая ее модификация, называемая глиоксилатным циклом (последовательность реакций глиоксилатного цикла представлена на рисунке 9).
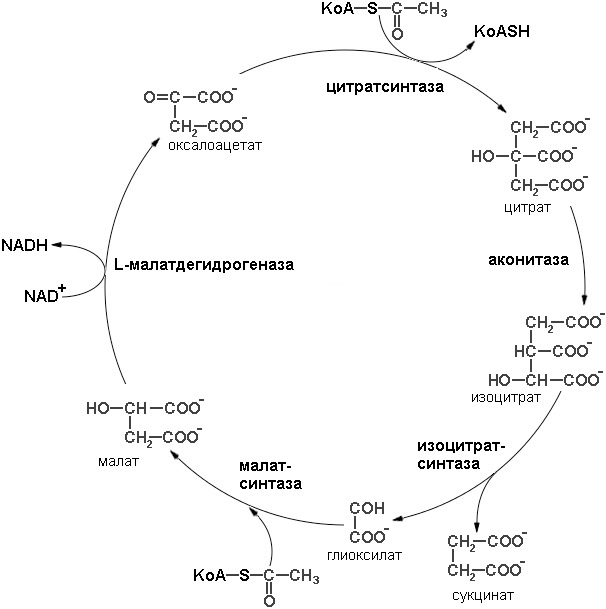
В глиоксилатном цикле ацетил-СоА взаимодействует с оксалоацетатом, в результате чего образуется цитрат. Однако расщепление изоцитрата происходит не в обычной изоцитратдегидроненазной реакции, как в цикле лимонной кислоты, а особым путем под действием фермента изоцитратлиазы с образованием сукцината и глиоксилата. Образовавшийся глиоксилат далее конденсируется с другой молекулой ацетил-СоА, что приводит к образованию малата, эта реакция катализируется малатсинтазой. Затем малат окисляется до оксалоацетата, который может конденсироваться с новой молекулой ацетил-СоА, начиная тем самым новый оборот цикла.
При каждом обороте глиоксилатного цикла в него вступают две молекулы ацетил-СоА и образуется одна молекула сукцината, которая затем используется в процессах биосинтеза. Сукцинат может превращаться через фумарат и малат в оксалоацетат из которого образуется фосфоенолпируват путем обращения описанной выше фосфоенолпируваткарбоксикиназной реакции. Фосфоенолпируват используется в качестве предшественника при биосинтезе глюкозы.
У животных глиоксилатный цикл отсутствует; изоцитратсинтазы и малатлиазы в животных клетках нет. В организме животных существуют другие пути для синтеза углеводов из простых предшественников.
В прорастающих семенах глиоксилатный цикл, напротив, функционирует очень активно: таким путем из ацетильных групп (источником которых служат жирные кислоты, входящие в состав запасных триацилглицеролов) образуется глюкоза. Ферменты изоцитратлиаза и малатсинтаза находятся в растительных клетках в особых цитоплазматических органеллах глиоксисомах.
Дыхательная цепь и окислительное фосфорилирование
Как было рассмотрено ранее, суммарная реакция окисления глюкозы до CO2 в реакциях гликолиза и цикле трикарбоновых кислот выглядит следующим образом:
Глюкоза +10NAD+ +2FAD+ +2АДФ+2 ГДФ = 6 CO2 +10NADH +2FADH2 +2ГТФ +2ATФ
Как видно из суммарной реакции энергетический выход данного процесса окисления невелик: 2 АТФ из гликолиза и 2 ГТФ из цикла трикарбоновых кислот, если цикл идет в печени, то есть только в печеночной ткани цикл трикарбоновых кислот дает энергетический выход в виде нуклеозидтрифосфатов. При этом основным продуктом окисления являются восстановленные доноры/акцепторы электронов или NADH и FADH2, поэтому важным фактором является доказательство возможности использовать данные молекулы в качестве источника энергии. Хотя в составе данных молекул присутствуют фосфоангидридные связи, характерные для нуклеозидтрифосфатов, и, следовательно, данные молекулы могут быть донорами энергии как АТФ, но гидролиз данных молекул не является экономически выгодным, так как затраты на синтез этих молекул слишком велик.
Вторая проблема связана с тем, что гидролиз связи может происходить как в окисленной, так и в восстановленной молекуле, следовательно, с такой точки зрения цикл трикарбоновых кислот становится бессмысленным, что для природы не характерно.
Из этого можно сделать вывод, что запасание энергии связано с процессом восстановления окислительно-восстановительных эквивалентов NAD+ и FAD+ и окислительно-восстановительными реакциями.
Основным параметром в окислительно-восстановительных реакциях является их способность отдавать (быть восстановителем) или принимать (быть окислителем) электроны в ходе реакции. Экспериментальной характеристикой этих способностей молекул является окислительно-восстановительный потенциал или red/ox потенциал.
Окислительно-восстановительные потенциалы и изменения свободной энергии
Окислительно-восстановительный потенциал — это электрохимическая категория. Необходимо рассмотреть для примера вещество, которое может существовать в окисленной X+ и в восстановленной форме Х. Такая пара называется окислительно-восстановительной парой (схема эксперимента для определения окислительно-восстановительного потенциала представлена на рисунке 10).
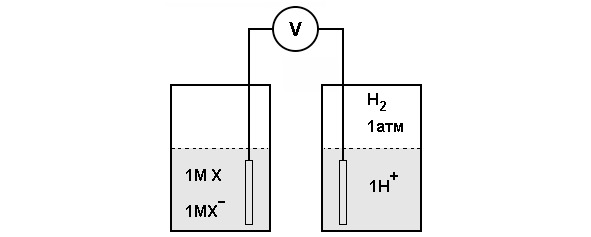
Окислительно-восстановительный потенциал такой пары можно определить, измеряя электродвижущую силу, развиваемую опытной полукамерой по отношению к стандартной контрольной полукамере. Опытная полукамера представляет собою электрод, погруженный в раствор 1 М окислителя (X+) и 1 М восстановителя (X). Стандартная контрольная полукамера состоит из электрода, погруженного в 1 М раствор Н+, находящийся в равновесии с газообразным Н2 при давлении в 1 атм. Электроды присоединяют к вольтметру и агаровым мостиком обеспечивают электропроводность между полукамерами. Происходит поток электронов oт одной полукамеры к другой. Если реакция идет в направлении
X + H+ → X+ +1/2H2.
то в полукамерах будут происходить следующие реакции:
Х → X+ + ē, H+ + ē → 1/2H2
Таким образом, электроны движутся от опытной полукамеры к контрольной и, следовательно, электрод в опытной полукамере заряжен отрицательно по отношению к электроду стандартной полукамеры. Окислительно-восстановительный потенциал пары X+:X соответствует напряжению в начале эксперимента (когда концентрации X+, X и Н+ равны 1 М). Окислительно-восстановительный потенциал пары Н+:Н2 определен равным 0 В (вольт).
Значение окислительно-восстановительного потенциала теперь очевидно. Отрицательный окислительно-восстановительный потенциал говорит о том, что данное вещество имеет меньшее сродство к электронам, чем Н2 (как в вышеприведенном примере), то есть молекула является донором электронов, то есть восстановителем. Положительный окислительно-восстановительный потенциал свидетельствует о более высоком, чем у Н2, сродстве данного вещества к электронам, то есть является окислителем или молекулой, с большей легкостью присоединяющей электроны. Эти соотношения относятся к стандартным условиям, когда концентрации окислителя, восстановителя и Н+ равны 1 М и давление Н2 составляет 1 атм.
Таким образом, сильный восстановитель (например, NADH) обладает отрицательным окислительно-восстановительным потенциалом, тогда как сильный окислитель (О2) имеет положительный окислительно-восстановительный потенциал. Окислительно-восстановительные потенциалы многих биологически важных окислительно-восстановительных пар известны.
Изменение свободной энергии окислительно-восстановительной реакции можно легко вычислить из разности окислительно-восстановительных потенциалов реагирующих соединений.
Любую окислительно-восстановительную реакцию в общем виде можно представить следующим образом:
Сначала нужно ввести обозначения: окислитель+ — окисленная форма окислителя, окислитель — восстановленная форма окислителя, восстановитель — восстановленная форма восстановителя, восстановитель+ — окисленная форма восстановителя. Запишем общую реакцию, испльзуя обозначения:
окислитель+ + восстановитель → окислитель +восстановитель+ (реакция А)
Любую окислительно-восстановительную реакцию разделяют на две полу-реакции, каждая из которых представляет собой обмен электронами между окисленной и восстановленной формами окислительно восстановительной пары, чей потенциал можно измерить в эксперименте, описанном выше:
окислитель+ + ē → окислитель (реакция Б)
Окислительно восстановительный потенциал этой пары Е1 восстановитель+ + ē→ восстановитель (реакция В)
Окислительно восстановительный потенциал этой пары Е2
Вычитая реакцию в) из реакции б), получаем желаемую реакцию а) и ΔE»о
Теперь можем рассчитать ΔG0» для восстановления пирувата за счет NADH. Изменение стандартной свободной энергии ΔG0» связано с изменением окислительно-восстановительного потенциала ΔE'о уравнением
ΔG0 = -nF ΔE»о
где n- число переносимых электронов, F-число Фарадея (23,062 ккал • В -1 • моль -1), ΔE'о выражается в вольтах, ΔG0 в килокалориях на моль.
Величина окислительно-восстановительного потенциала дыхательной цепи составляет 1,14 В, что соответствует 53 ккал.
Изменение окислительно-восстановительного потенциала при переходе от системы NAD+/NADH к системе О2/Н2О составляет 1,1 В.
Движущая сила окислительного фосфорилирования — это потенциал переноса электронов, присущий NADH или FADH2. Рассчеты ΔE'о и ΔG0 связанные с окислением NADH под действием О2. Промежуточные частичные реакции следующие:
а) 1/2О2 +2Н+ +2 ē→ Н2О
E«о = +0,82 В,
б) NAD+ + Н+ +2 ē→ NADH
E«о = — 0,32 В.
Вычитая реакцию б) из реакции а), получаем
в) 1/2 О2 + NADH + Н+ → Н2О + NAD +
ΔE'о = +1,14 В.
Свободная энергия окисления для этой реакции составляет
ΔG0 = -2—23,062.1,14 = — 52,6 ккал/моль.
Таким образом доказано, что молекула NADH является источником энергии, и, как показывают расчеты, при окислении этой молекулы с участием кислорода выделяется такое количество энергии, которое достаточно для синтеза 7 молекул АТФ. Но реакция происходит взрывообразно, и это не позволяет перевести энергию в более адекватную форму.
Чтобы обеспечить перевод энергии окисления в энергию АТФ необходима система окисления, это обеспечивает дыхательная цепь, состоящая из 4-х белковых комплексов, содержащих коферменты, участвующие в окислительно-восстановительных реакциях. В результате мы имеем с одной стороны материальную группу молекул, передающих электроны друг от друга, то есть образуется система передачи электронов от NADH к О2 по которому идут электроны, как электрическая цепь в сети, с другой стороны это последовательность окислительно-восстановительных реакций, которые происходят в составе электронтранспортной цепи, молекулы коферментов являются окислителями (акцепторами электронов) при взаимодействии с предшествующими молекулами, и являются восстановителями (донорами электронов) при взаимодействии со следующей молекулой цепи.
Из этого заключаем, что каждый следующий кофермент является большим окислителем, чем предыдущий, то есть этот кофермент «отбирает» электроны у предыдущего кофермента, а у него электроны «отбирает» следующий кофермент в электронтранспортной цепи. В данном случае наблюдается увеличение окислительно-восстановительного потенциала, следовательно, каждый следующий кофермент в электронтранспортной цепи является большим окислителем, и, как следствие, электронтранспортная цепь не только физическая часть для потока электронов, но и последовательность окислительно-восстановительных реакций.
Организация электронтранспортной цепи
Электронтранспортная цепь организована во внутренней мембране митохондрий и представляет собой четыре белковых комплекса, содержащих коферменты, окислительно-восстановительных реакций (общий план организации и функционирования электрон-транспортной цепи изображен на рисунке 11).
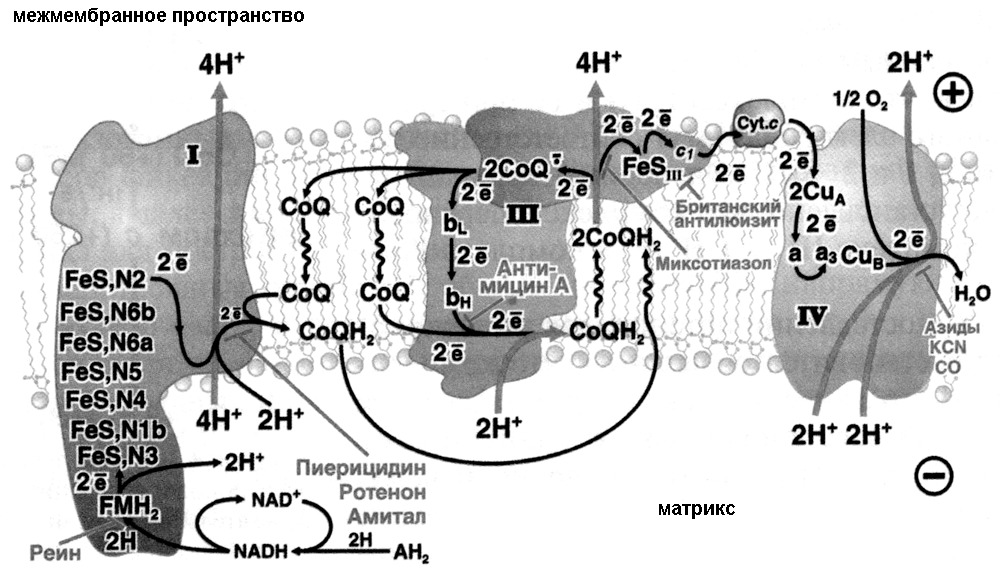
Первый комплекс — NADH-дегидрогеназа (комплекс I), сукцинат-дегидрогеназа (комплекс II), убихинон-цитохром с редуктаза (комплекс III), цитохром оксидаза (комплекс IV).
NADH-дегидрогеназа (комплекс I) включает в себя флавинмононуклеотид (FMH) и как минимум шесть Fe-S комплексов. Известны три вида Fe-S-центров. В простейшем случае единственный атом железа тетраэдрически координирован с сульфогидрильными группами четырех цистеиновых остатков белка. Второй вид комплексов (обозначен как Fe2-S2) содержит 2 атома железа и два неорганических дисульфида, присоединенных к четырем цистеиновым остаткам. В комплексах третьего вида (Fe4-S4) содержится четыре атома железа, четыре неорганических сульфида и четыре остатка цистеина. В состав NADH-дегидрогеназы входят два кофермента класса Fe2-S2, и четыре класса Fe4-S4 (Схема организации и функционирования коферментов комплекса I представлена на рисунке 12).
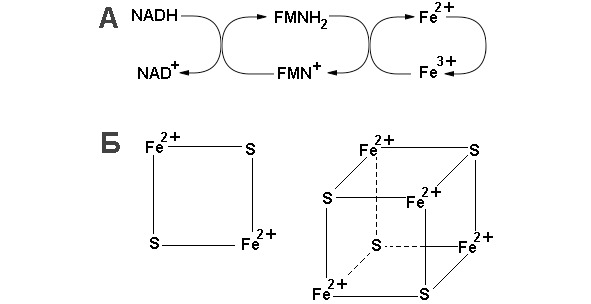
Сукцинат-дегидрогеназа (комплекс II) состоит из 4 субъединиц с молекулярными массами 70, 30, 14 и 12 кDa и содержит в качестве окислительно-восстановительных групп флавинадениндинуклеотид (FAD), ковалентно связанный с самой тяжелой субъединицей, и 3 Fe-S-кластера (один Fe2-S2 и два Fe4-S4), ассоциированных с субъединицей с молелекулярной массой 30 кDa.
Комплекс I обеспечивает окисление молекулы NADH, а комплекс II окисляет молекулу FADH2, электроны поступают на коферменты электрон-транспортной цепи и в конце концов поступают на молекулу убихинона или кофермента Q. Кофермент Q — хиноновое производное с длинным изопреноидным хвостом. Его называют также убихиноном из-за его повсеместного распространения в биологических системах.
Число изопреновых единиц в коферменте Q зависит от вида живых организмов, У млекопитающих его наиболее распространенная форма содержит десять изопреновых единиц и обозначается как Q10. Изопреноидный хвост обуславливает высокую неполярность Q, которая способствует его быстрой диффузии в углеводородной фазе внутренней митохондриальной мембраны. Кофермент Q является компонентом митохондриальных липидов; среди других липидов преобладают фосфолипиды, являющиеся частью митохондриальной мембраны. Структура кофермента Q сходна со структурой витаминов К и Е.
Близкую структуру имеет и пластохинон, находящийся в хлоропластах. Все эти вещества имеют в своей структуре полиизопреноидную боковую цепь. Содержание кофермента Q значительно превосходит содержание других компонентов дыхательной цепи (по параметру стехиометрии); это позволяет предположить, что кофермент Q является подвижным компонентом дыхательной цепи, который получает восстановительные эквиваленты от фиксированных флавопротеиновых комплексов и передает их на цитохромы. Кофермент Q-единственный переносчик электронов в дыхательной цепи, который не связан прочно с белком и не присоединен к нему ковалентно. Кофермент Q действительно служит высокомобильным переносчиком электронов между флавопротеинами и цитохромами цепи переноса электронов.
Убихинон-цитохром с редуктаза (комплекс III) включает 11 субъединиц с молекулярным весом: 49,5, 47, 44, 28, 21,5, 13,5, 9,5, 9, 8, 7, 6,5 kDa соответственно. Третья субъединица весом 44 kDa присоединяет две молекулы гемов bH и bL. Центральную роль цитохромов в дыхании открыл в 1925 г. Дэвид Кейлин (David Keilin).
Цитохром — это переносящий электроны белок, молекула которого содержит в качестве простетической группы гем.
Гем — это модифицированная молекула тетрапиррольного кольца, в центре которой ассоцииирован ион металла (это могут быть ионы железа, меди и других металлов). В зависимости от радикалов, модифицирующих кольцо, и от ионов ассоцированных с кольцом гема, выделяют несколько классов цитохромов. Субъединица V или белок Риске содержит Fe2-S2 кластер. Субъединица VI связывает убихинон, субъединица IV координирует цитохром с. Функции остальных субъединиц не выявлены или участвуют в организации комплекса (схема функционирования убихинона и коферментов комплекса III представлена на рисунке 13).
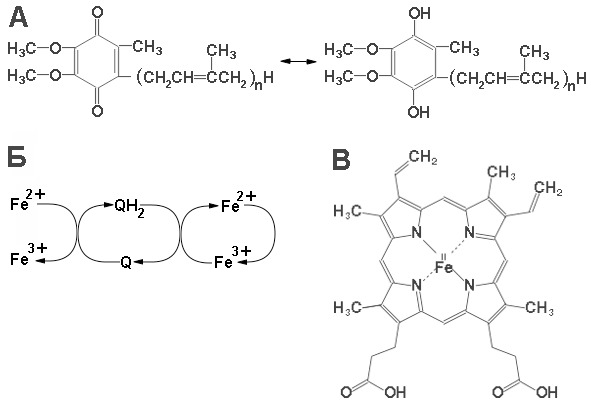
Электроны с убихинон-цитохром с редуктазы переносятся на цитохром с. Цитохром с — водорастворимый, подвижный белок с молекулярной массой 12 kDa. Этот белок мигрирует между комплексами III и IV в межмембранном пространстве.
Цитохром оксидаза (комплекс IV) содержит восемь белковых субъединиц, с ними ассоциированы два гема, содержащих ионы меди, которые называют гемы а и а3. Кроме этого содержит два иона меди CuA и СuB. Центр СuB представляет ион меди соединенный с радикалами трех остатков гистидина. Центр CuA содержит два атома меди расположенных очень близко и скоординированных с белком (схема функционирования коферментов комплекса IV представлена на рисунке 14).
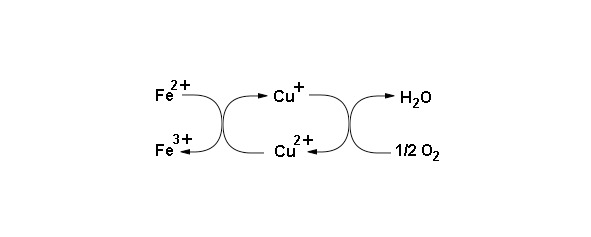
Механизмы переноса электронов в электронтранспортной цепи
Первый комплекс или NADH-дегидрогеназа взаимодействует с молекулой NADH, электроны переносятся на (FMN), в результате FMN принимает с NADH два электрона и два протона от среды, и образуется NAD+ и FMNH2, затем электроны переносятся на первый комплекс Fe4-S4 белка в результате происходит окисление FMNH2 и перенос электронов на ион железа Fe-S белка, далее железо в комплексе восстанавливается из положения Fe3+ до Fe2+. Это происходит потому, что окислительно-восстановительный потенциал NADH меньше, чем у FMN, поэтому происходит окисление NADH флавинмононуклеотидом, который затем окисляется Fe4-S4 белком, чей окислительно-восстановительный потенциал еще больше. Далее происходит серия окислительно-восстановительных реакций с участием Fe-S белков, каждый из которых является большим окислителем, чем предыдущий.
В результате восстановленный ион железа Fe2+ Fe-S белка предшественника, окисляется до положения Fe3+, следующим цепи транспорта электронов Fe-S белком, чей окислительно-восстановительный потенциал выше, чем у предшественника, в результате ион железа окислителя восстанавливается из положения Fe3+ с Fe2+.
Особенностью флавинмононуклеотида является способность переносить два электрона, как и другие нуклеотидсодержащие коферменты, тогда как железо-серные белки переносят только по одному электрону, именно поэтому флавинмононуклеотид является адаптором процесса электронов с NADH на Fe-S белки.
Затем происходит последовательный перенос с одного Fe-S белка на другой, где один ион железа окисляется, а второй восстанавливается. Точно также ион железа Fe-S белка восстановителя окисляется из Fe2+ в Fe3+, а в белке окислителе восстанавливается из Fe3+ в Fe2+. Конечным акцептором является Fe4-S4 белок, чья восстановленная Fe2+ форма, окисляется до Fe3+ убихиноном, который обладает большим окислительно-восстановительным потенциалом. В результате окисленная форма хинона Q переходит в восстановленную QH2.
Вторым источником электронов для убихинона является сукцинатдегидрогеназа: фермент цикла трикарбоновых кислот. Два электрона переносятся с сукцината на FAD в цикле трикарбоновых кислот. В результате FAD восстанавливается до FADH2, который окисляется Fe-S-белками, каждый из которых является большим окислителем и точно также происходит перенос электронов на ион железа в Fe-S-белках, а в конце концов на убихинон, который в результате восстанавливается до QH2.
Восстановленный убихинон (QH2) диффундирует к комплексу III, где связывается с субъединицей QIII, где происходит окисление восстановленного убихинона, протоны высвобождаются в межмембранное пространство, а электроны поступают на электронтранспортную цепь.
Один электрон поступает на на ген bL, а затем гем bH, а потом на хинон для дальнейшего восстановления за счет работы комплекса I или комплекса II. А вот второй электрон поступает на Fe-S белок, в результате происходит восстановление иона железа из Fe3+ до Fe2+, так как этот комплекс более сильный окислитель. Далее Fe-S белок окисляется цитохромом с1, содержащим в составе гема ион железа. В ходе реакции ион железа в Fe-S белке окисляется от Fe2+ в Fe3+, а в цитохроме с1 ион железа восстанавливается Fe3+ до Fe2+. Затем цитохром с1 окисляется подвижным цитохромом с, также содержащим ион железа, который восстанавливается до положения Fe2+.
Цитохром с мигрирует к последнему комплексу IV (цитохромоксидазе). Взаимодействуя с цитохромоксидазой цитохром с окисляется ионом меди в комплексе CuA. В результате ион железа в цитохроме с окисляется от Fe2+ в Fe3+ а ион меди в CuA восстанавливается Cu2+ в Cu+. В свою очередь комплекс окисляется ионом железа в геме а. А ион железа в составе гема а окисляется ионом железа в геме а3, который востанавливается.
Восстановленный ион железа в геме а3 окисляется ионом меди в комплексе CuB. Ион меди в этом в этом комплексе восстанавливается, а затем окисляется кислородом, параллельно присоединяя четыре протона, с образованием двух молекул воды.
Таким образом происходит перенос электронов от NADH к кислороду, то есть потока электронов или цепи последовательных окислительно-восстановительных реакций, где почти каждая молекула является акцептором электронов в предшествующей реакции и донором в последующей.
Как было рассмотрено выше, в ходе окислительно-восстановительных реакций также выделяется и поглощается энергия, в начальных этапах исследований считалось, что выделившаяся энергия затрачивается на синтез некоего макроэргического соединения, которое, гидролизуясь, дает энергию для фосфорилирования АДФ и образования АТФ. Но дальнейшие исследования показали, что на самом деле никакого соединения нет. А все экспериментальные исследования подтвердили гипотезу, а затем теорию предложенную в 1961 году Питером Митчеллом.
Хемиосмотическая гипотеза Митчелла
Митчелл предположил, что сопряжение переноса электронов и синтеза АТФ обеспечивается протонным градиентом, а не высокоэнергетическим ковалентным промежуточным продуктом или активированным белком. Согласно этой модели, перенос электронов по дыхательной цепи приводит к выбросу протонов из матрикса на цитоплазматическую сторону внутренней митохондриальной мембраны, где, таким образом, возрастает концентрация ионов Н+. В результате происходит генерирование мембранного потенциала с положительным зарядом на цитоплазматической стороне мембраны.
Гипотеза Митчелла о сопряжении окисления и фосфорилирования протонным градиентом получила к настоящему времени множество подтверждений.
1. Согласно Митчеллу, первичным событием в окислительном фосфорилировании является транслокация протонов (Н+) на наружную сторону сопрягающей мембраны (внутренней митохондриальной мембраны), осуществляемая за счет процесса окисления в дыхательной цепи. При этом предполагается, что мембрана непроницаема для ионов вообще, особенно для протонов, которые накапливаются на наружной стороне мембраны, создавая по обе стороны мембраны разность электрохимических потенциалов (Δμн). Она складывается из химического потенциала (разность рН) и электрического потенциала.
Разность электрохимических потенциалов обеспечивает действие локализованной в мембране АТФ-синтазы (или обращение процесса, катализируемого локализованной в мембране АТФ-гидролазой), которая в присутствии Фн -+ AДФ синтезирует ATФ. Таким образом, нет необходимости в высокоэнергетическом промежуточном соединении, общем для процессов окисления и фосфорилирования, как это постулирует химическая гипотеза.
Протонный градиент через внутреннюю митохондриальную мембрану создается во время переноса электронов. рН с наружной стороны на 1,4 единицы ниже, чем с внутренней, и мембранный потенциал составляет 0,14 В, причем наружная сторона несет положительный заряд. Общий электрохимический потенциал Δр (в вольтах) складывается из мембранного потенциала (ΔΨ) и градиента концентрации ионов Н + (ΔрН). В приведенном ниже уравнении R-газовая постоянная, Т — абсолютная температура, F- число Фарадея.
Δр = Δψ — RT/F*ΔрН
как видно из уравнения в него входят два компонента электрический (Δψ) и химический (RT/F*ΔрН). Электрический компонент связан с запасанием энергии, в данном случае наблюдается следующая картина:, за счет переноса протонов, водный раствор межмембранного пространства заряжен положительно, а водная часть матрикса митохондрии наоборот отрицательно, а мембрана за счет гидрофобного билипидного слоя выступает в роли прослойки из диэлектрика (это похоже на две заряженные пластины, разделенные слоем диэлектрика — а это уже конденсатор). В случае разницы в концентрациях протонов между двумя указанными выше компартментами — это тоже форма запасания энергии. Известно, что на создание градиентов затачивается энергия АТФ, например работа К/Na-АТФ-азы. При подстановке результатов, полученных при измерении, в уравнение можно получить результаты приведенные ниже.
Δр = Δψ — RT/F*ΔрН = Δψ — 0,06ΔрН = 0,14 — 0,06 (- 1,4) — 0,224 В.
Эта общая протонодвижущая сила в 0,224 В соответствует свободной энергии 5,2 ккал в расчете на 1 моль протонов. Следовательно для синтеза одного моля АТФ необходим перенос 2 моль протонов.
2. При создании градиента рН в митохондриях или хлоропластах в них происходит синтез АТР в отсутствие переноса электронов.
3. Белок пурпурных мембран галобактерий при освещении перекачивает протоны. Синтетические пузырьки, содержащие этот бактериальный белок и очищенную АТР-азу из митохондрий сердца крупного рогатого скота, синтезируют АТФ при освещении. В этом опыте белок пурпурных мембран заменяет дыхательную цепь; следовательно, дыхательная цепь и АТФ-аза — биохимически отдельные системы, связываемые только протонным градиентом.
4. И дыхательная цепь, и АТФ-аза имеют векторную организацию во внутренней митохондриальной мембране.
5. Для окислительного фосфорилирования существенное значение имеет замкнутость компартментов. В растворимых препаратах или в мембранных фрагментах, лишенных хорошо отграниченных внутренних и внешних компартментов, не происходит синтеза АТФ, сопряженного с переносом электронов.
6. Вещества, переносящие протоны через внутреннюю митохондриальную мембрану, разрушают протонный градиент и таким образом вызывают разобщение окисления и фосфорилирования.
7. Компоненты дыхательной цепи уложены в мембране упорядоченно, «бок о бок», поперек мембраны, как предусматривается хемиосмотической теорией.
Второй важный факт это точки переноса протонов, или точки генерации протонного градиента, так как естественно, что в каждой реакции генерация не происходит. Следовательно важным моментом является определение количества и положения точек генерации протоннного градиента.
Механизмы генерации протонного градиента
Согласно хемиосмотической теории энергия окислительно-восстановительных реакций тратится на перенос протонов из матрикса митохондрии в межмембанное простанство. Но не все реакции в электронтранспортной цепи вызывают перенос протонов. Эти реакции выявлялись несколькими методами.
1. Соотношение Р/О при окислении нескольких субстратов.
В данном случае изучают соотношение фосфата, перешедшего в состав органичеких соединений к поглощенному кислороду. Для этого измеряют концентрацию свободного фосфата в среде с митохондириями до добавления субстрата, а затем после. Полученная разность концентраций является количеством фосфата, который перешел из свободного в связанное состояние, то есть был затрачен на синтез АТФ из АДФ и фосфата. Второй параметр — это поглощенный кислород. Измеряем концентрацию кислорода в начале и в конце опыта, разность равна количеству кислорода превратившемуся в воду в ходе работы электронтранспортной цепи. Делим разность концентраций свободного фосфата в среде на разность содержания кислорода и получаем соотношение P/O. Это значение различается для разных субстратов добавленных в оытную смесь с митхондриями.
2. Термодинамические измерения.
Для каждой реакции в электронтранспортной цепи можно рассчитать изменение свободной энергии (ΔG0), так как известны окислительно-восстановительные потенциалы для каждой полупары в реакции. Получив значения ΔG0, для каждой реакции можно предположить, выделяется ли достаточное количество энергии для переноса протонов. Значения во всех реакциях меньше нуля, так как каждая реакция идет самопроизвольно. Но выделившаяся энергия не всегда достаточна для переноса протонов. Если значение модуля ΔG0 превышает модуль значения ΔG0 в реакции гидролиза АТФ, тогда выделившейся энергии достаточно для реакции фосфорилирования АДФ до АТФ. Таким образом, все реакции электронтранспортной цепи, значение модуля ΔG0 которых больше чем модуль ΔG0 реакции гидролиза АТФ подходят для генерации протонного градиента.
3. Специфическое ингибирование тока электронов.
Места действия этих ингибиторов были установлены с использованием метода перекреста. Бриттон Чане предложил изящный спектроскопический метод для определения соотношения окисленной и восстановленной форм каждого переносчика. В основе метода лежит тот факт, что окисленная и восстановленная формы каждого переносчика имеют свои характерные спектры поглощения. Добавление ингибитора переноса электронов изменяет соотношение этих форм. Например, добавление антимицина А вызывает переход переносчиков, локализованных в электронтранспортной цепи между NADH и цитохромом b, в более восстановленное состояние, а переносчиков между цитохромом с и О2 в более окисленное состояние. Отсюда можно заключить, что антимицин А подавляет превращение цитохрома b в цитохром ci, потому что этот этап является пунктом перекреста.
Перенос протонов осуществляется за счет активности белков переносчиков, но возможны два пути: первый — это «Митчелова петля», когда перенос электронов сопряжен с переносом протонов, то есть реакции сопряжены, второй путь — это протонная помпа, когда окислительно- восстановительная реакция является только источником энергии для переноса протонов.
Таким образом можно рассмотреть механизмы генерации протонного градиента белковыми комплексами, входящими в состав электронтранспортной цепи.
Первая точка генерации протонного градиента располагается в NADH-дегидрогеназном комплексе (комплекс I), между Fe-S белками чей потенциал значительно выше чем у FMN, и в результате реакции выделяется достаточное количество энергии для переноса четырех протонов из матрикса митохондрии в межмембранное пространство, все исследования позволяют предполагать, что перенос осуществлялся по механизму протонной помпы.
В ходе активности сукцинатдегидрогеназы (комплекса II) перенос протонов не осуществляется.
В работе убихинон-цитохром с -редуктазы (комплекса III) также переносятся четыре протона. Это происходит параллельно c переносом электронов по электрон-транспортной цепи. Первый электрон переносится на [Fe — S] кластер белка Риске, а второй электрон передается на низкопотенциальный гем bL цитохрома b. Дальнейшая судьба этих электронов оказывается различной. Электрон с [Fe — S] кластера переносится на цитохром сb далее — на цитохром с и цитохром с-оксидазу. В то же время электрон с тема bL переносится на высокопотенциальный гем Ьн и далее используется для восстановления молекулы хинона в центре N с образованием семихинона. При окислении в центре Р второй молекулы хинона еще два протона высвобождаются в межмембранное пространство митохондрий, образуется еще одна молекула восстановленного цитохрома с, а убисемихинон в центре N восстанавливается до хинола. Для образования хинола в ценре N два протона захватываются из матрикса митохондрий. Образованный в центре N хинол отсоединяется от фермента и диффундирует на другую сторону мембраны, чтобы снова окислиться в центре Р.
Перенос электрона от центра N к цитохрому с происходит вдоль мембраны, поэтому этот процесс не приводит к запасанию энергии. А вот перенос электрона от центра Р к центру N является трансмембранным, что ведет к генерации электрического потенциала. Таким образом, генерация протонного потенциала комплексом III происходит с использованием механизма Митчеловой петли.
Редокс-потенциалы переносчиков электронов, присутствующих в СорНг-цитохром с-редуктазе, хорошо согласуются со схемой Q-цикла. Для гемов bL и bн они равны соответственно -40 и +40 мВ, для [Fe-S] кластера +280, а для цитохромов и с — соответственно +220 и +250 мВ. Важно, что редокс-потенциал гема Ън заметно положительней, чем редокс-потенциал гема bL, так что перенос электронов с bL на Ьн должен сопровождаться высвобождением энергии. Эта энергия запасается в виде ДЧ*, поскольку оксидоредукция bH^> bL направлена поперек мембраны. Перенос электронов от [Fe — S] Риске к цитохрому сх и далее к с вдоль мембраны происходит без существенных изменений редокс-потенциала и, следовательно, без выделения энергии.
Суммарная реакция реакция комплекса III может быть записана в следующем виде:
2CoQH2 +2цит. с (0Х) + CoQ +2H+in → 2CoQ +2цит. c (red) +CoQH2+4H+out +2q.
Или после сокращения:
CoQH2 +2цит. с (ох) +2Н+in → CoQ +2цит. c (red) +4H+out +2q.
Таким образом, перенос двух электронов с убихинола на цитохром с сопровождается захватом двух протонов из матрикса митохондрий, выбросом четырех протонов в межмембранное пространство и переносом двух зарядов
В ходе активности цитохром оксидазы (комплекса IV) с цитохрома а на диядерный центр, связанный с кислородом проиходит перенос двух протонов по механизму «Митчеловой петли». По другим данным этот же процесс происходит по механизму протонной помпы.
Как было показано в результате исследований в ходе функционироания электронтранспортной цепи при окислении NADH переносится 10 протонов, а при окислении FADH2 происходит перенос шести протонов.
Однако количество переносимых протонов, не совсем соответствует тем данным, которые есть во всех учебниках, и этот факт требует разъяснения. Количество АТФ, синтезированного в смеси интактных митохондрий и окисляемого субстрата, например сукцината или NADH, в присутствии кислорода оценивалось экспериментально по уменьшению концентрации кислорода. Однако измерить коэффициент P/О было трудно, поскольку интактные митохондрии используют АТФ в различных биохимических процессах, а кислород расходуется не только на окислительное фосфорилирование. Поэтому в большинстве таких экспериментов для P/О получали дробные числа, например в случае NADH значения этих коэффициентов были в интервале между 2 и 3, в случае сукцината — между 1 и 2. Тем не менее в течение многих лет для величины P/О были приняты целочисленные значения, равные 3 и 2 для NADH и сукцината соответственно.
Хемиосмотическая теория, объясняющая механизм сопряжения переноса электронов в дыхательной цепи митохондрий с синтезом АТФ в митохондриях, не постулирует целочисленность коэффициента P/О, а подходит к вопросу о стехиометрии процесса окислительного фосфорилирования с иной позиции. Согласно хемиосмотической теории, для оценки стехиометрии процесса окислительного фосфорилирования, т. е. величины P/О, необходимо учитывать не только число протонов, выкачиваемых из матрикса через внутреннюю мембрану митохондрии за счет энергии, высвобождающейся при переносе электронов от одной молекулы NADH к кислороду, но и число протонов, вернувшихся в матрикс через протонный канал в комплексе F0F1, в результате чего появляется движущая сила для синтеза АТA.
Бесплатный фрагмент закончился.
Купите книгу, чтобы продолжить чтение.